Стратегия лечения направлена на коррекцию метаболических нарушений комплексом мероприятий: консервативными методами (патогенетическая диетотерапия, левокарнитин), хирургическими методами (лечение внутричерепных кровоизлияний и костных деформаций), реабилитационными методами. Лечение должно быть начато всем больным с выявленным достоверным повышением уровня глутаровой кислоты в моче до получения результатов ДНК диагностики ГА1. Для лечения энцефалитоподобного криза в международные стандарты включены методы интенсивной терапии. Также проводится симптоматическая терапия с тщательным подбором лекарственных препаратов, направленных на коррекцию неврологических нарушений.
3.1 Консервативное лечение
- Рекомендуется патогенетическая диетотерапия с исключением высокобелковых продуктов, богатых лизином и триптофаном, обязательным использованием специализированных продуктов на основе смесей аминокислот без указанных патогенетически значимых аминокислот пациентам с установленным диагнозом ГА1 с целью предотвращения образования нейротоксических метаболитов [1, 3, 7, 18, 19, 20].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: Диетотерапия с низким содержанием лизина и триптофана должна соблюдаться строго пожизненно. Диетотерапия при заболевании направлена на снижение поступления лизина, триптофана, которые являются основными предшественниками нейротоксических метаболитов - глутаровой и 3-OH-глутаровой кислот. Данные аминокислоты содержатся в большом количестве в белках животного происхождения (рыбе, мясе, молочных продуктах и др.). В среднем содержание в них лизина составляет 6 - 9% и триптофана 0,6-2%. В растительных же продуктах (овощи, фрукты, некоторые крупы) содержание указанных аминокислот минимально.
При ГА 1 применяют два варианта лечебного питания: первый - диета с низким содержанием лизина и триптофана с использованием специализированного продукта, данный вариант диеты показан в первую очередь детям (с рождения до 18 лет), у взрослых пациентов также может быть использован этот вариант диеты, но и можно применять второй - лечебный рацион с низким содержанием общего белка.
Пациенты первого года жизни могут находиться на грудном вскармливании. Грудное молоко в данном случае будет являться основным источником натурального белка в сочетании со специализированной смесью аминокислот, по составу соответствующей возрасту ребенка. Количество лизина в грудном молоке известно и составляет примерно 86 мг на 100 мл.
Применение второго варианта диеты повышает риск алиментарной недостаточности незаменимых аминокислот и других эссенциальных нутриентов, что может привести к развитию различных нарушений: повышенной возбудимости, нарушению сна, немотивированным колебаниям температуры, пеллагре (в результате снижения образования никотиновой кислоты), именно поэтому данный вариант может быть применен только у пациентов старшего возраста. Специализированные продукты лечебного питания на основе аминокислот без лизина и триптофана**** представлены в Приложении А6.
Минимально допустимые потребности патогенетически значимых аминокислот в питании детей различного возраста представлены в Приложении А7 [1, 2, 18].
Пациенты младенческого возраста могут находиться на грудном вскармливании. Грудное молоко в данном случае будет являться основным источником натурального белка в сочетании со специализированной смесью аминокислот, соответствующей возрасту ребенка. Количество лизина в грудном молоке известно и составляет 86 мг на 100 мл.
При расчете низкобелковой диеты со сниженным содержанием лизина и триптофана необходимо помнить, что лизин и триптофан содержатся практически во всех продуктах, и учитывать это при составлении лечебного рациона.
Специализированная диета требует мониторинга за уровнем аминокислот и их метаболитов в плазме крови и моче, биохимических маркеров нутритивного статуса (общий белок, протеинограмма и др.), динамического наблюдения за физическим развитием и нутритивным статусом пациента, а также периодической коррекции лечебного рациона с расчетом его химического состава.
В приложении А8 представлены таблицы, в которых содержатся международные и российские рекомендации по суточным нормам потребления белка, лизина и триптофана и данные по среднему содержанию лизина в некоторых натуральных продуктах.
3.2 Медикаментозная терапия
- Рекомендуется пожизненно патогенетическая терапия #левокарнитином пациентам с ГА1 с целью выведения токсичных метаболитов и коррекции вторичной недостаточности карнитина [1, 2, 7, 16, 19, 20, 65].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
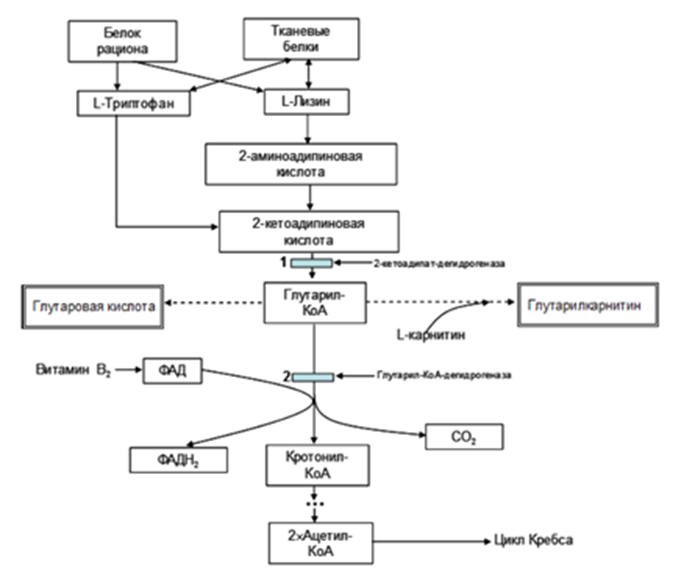
Комментарии: при ГА1 наблюдается вторичная недостаточность карнитина в тканях. Его пул быстро истощается, поскольку он связывается с глутаровой кислотой, которая присутствует у пациентов в больших количествах. Препараты, содержащие Левокарнитин восполняют этот вторичный дефицит, связывют глутаровую кислоту и обеспечивают ее выведение из организма в виде глутарилкарнитина. У большинства нелеченых пациентов с ГА 1 концентрация общего и свободного карнитина в плазме и сыворотке крови низкая. Начальная доза #Левокарнитина составляет 100 мг/кг/сутки для всех возрастов, она может быть снижена до 30мг/кг/сутки у пациентов старше 6 лет при нормальном уровне свободного карнитина в крови [20, 65].
В Приложении А9 суммированы основные мероприятия, которые обязательно должны быть выполнены после подтверждения диагноза ГА1.
3.3 Хирургическое лечение
- Рекомендуется хирургическое лечение субдуральных гидром и арахноидальных кист, гидроцефалии и других патологий пациентам с ГА1 по показаниям с целью стабилизации состояния при нейрохирургической патологии [7, 19, 20].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: Решение о целесообразности проведения нейрохирургического вмешательства (вентрикулоперитонеальное шунтирование и эвакуация субдуральных скоплений в мозге при прогрессирующей макроцефалии) принимается в каждом конкретном случае консилиумом врачей специалистов. Нейрохирургическая терапия арахноидальных кист и субдуральных гематом у пациентов с ГА1 должна проводиться крайне осторожно под наблюдением детского нейрохирурга.
- Рекомендуется хирургическое лечение костных деформаций (тяжелые контрактуры, вывих бедра и др.) пациентам с ГА1 по показаниям с целью стабилизации и улучшения состояния при ортопедической патологии [2, 7, 39].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 4)
Комментарии: Решение о целесообразности проведения нейроортопедического вмешательства принимается в каждом конкретном случае консилиумом врачей специалистов с соответствующей коррекцией метаболических нарушений с целью предотвращения развития криза.
- Рекомендуется при планировании хирургического вмешательства по возможности избегать голодания, применять инфузии декстрозы** и удвоить дозировку левокарнитина [21].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: Решение о целесообразности проведения нейроортопедического вмешательства принимается в каждом конкретном случае консилиумом врачей специалистов. Назначение двойной дозы левокарнитина в соответствии с инструкцией- вторичная недостаток карнитина или его повышенной потерей.
3.4 Иное лечение
Лечение неврологических состояний проводится согласно соответствующим клиническим рекомендациям (неврологические состояния).
- Рекомендуется при возникновении лихорадки выше 38,5 назначать ибупрофен**или парацетамол**[28].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: иные НПВС могут вызвать синдром Рейе.
- Рекомендуется при возникновении инфекционных и интеркуррентных заболеваний назначать лечение согласно соответствующим клиническим рекомендациям (инфекционные заболевания). Антибактериальную терапию назначают в зависимости от предполагаемого патогенного агента в возрастных дозах [21].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5)
Комментарии: противопоказания к назначению любых групп антибактериальных препаратов отсутствуют.
- Рекомендуется фармакотерапия экстрапирамидных нарушений препаратами из группы миорелаксантов центрального действия, пациентам с ГА1 по показаниям с целью коррекции неврологический нарушений [2,16,21, 55].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5)
Комментарии: Препаратами первой очереди для коррекции спастичности и мышечной дистонии является баклофен** и препараты из группы производные бензодиазепина (диазепам**, клоназепам**), направленные на коррекцию генерализованных и фокальных дистоний. Доза препаратов подбираются индивидуально согласно общепринятым стандартам терапии неврологических нарушений. Интратекальное применение баклофена** может быть использовано при выраженных дистониях и спастичности.
Препаратом второй очереди при выраженном дистоническом синдроме считаются препараты леводопы (#Леводопа+[Карбидопа]**, #Леводопа+[Бенсеразид]**) и #Тригексифенидил**, который назначается детям в возрасте старше 5 лет [55, 67, 68].
Общепринятый принцип назначения #Тригексифенидила** у детей с дистонией: стартовать с низкой дозы с постепенным повышением каждую неделю до уровня приблизительно 1-2 мг/кг, разделенных на 3 приема. Некоторым пациентам могут понадобиться более высокие дозы. Длительность лечения определяется индивидуально – от нескольких недель до нескольких месяцев [59]. Начальная доза #тригексифенидила** не должна превышать 0,5–1 мг/сут.
Дозировки #Тригексифенидила** у детей с дистонией согласно проведенным исследованиям:
1-я неделя: 0,05 мг/кг х2р/сут
2-я неделя: 0,05 мг/кг х3р/сут
3-я неделя: 0,1 мг/кг х3р/сут
4-я неделя: 0,15 мг/кг х3р/сут
5-я неделя: 0,20 мг/кг х3р/сут
6,7,8,9 недели – 0,25 мг/кг х3р/сут [60].
или
1-я неделя - 0,2 мг/кг/сут – разделенные на 3 приема
2-я неделя - 0,5 мг/кг/сут – разделенные на 3 приема
3-я неделя – 1,0 мг/кг/сут – разделенные на 3 приема
4-я неделя – 1,5 мг/кг/сут – разделенные на 3 приема
5-я неделя - 2,0 мг/кг/сут – разделенные на 3 приема
6-я неделя – 2,5 мг/кг/сут – разделенные на 3 приема
7-12-я недели – 2,5 мг/кг/сут – разделенные на 3 приема [61].
У взрослых с дистонией #тригексифенидил** обычно начинают с дозы 2мг х 2-3 р/сут с постепенным повышением на 2мг каждую 1-2 недели (чаще каждые 2 недели), обычно до достижения 21,5 мг [62].
Применение антихолинергических средств следует ограничивать в связи с риском появления или усугублении когнитивных и психических нарушений [40,63].
Следует учитывать, что выраженная спастичность, гиперкинезы и дистонические атаки могут приводить к выраженному болевому синдрому и белково-энергетической недостаточности, которые, в свою очередь, провоцирует усиление двигательных нарушений. Таким образом, в комплекс лечения спастичности и дистоний обязательно должны входить мероприятия для предотвращения и купирования болевого синдрома и белково-энергетического дефицита, с учетом диетических ограничений [41].
- Рекомендуется ботулинотерапия пациентам с ГА1 по показаниям с целью коррекции неврологический нарушений (дистонии, спастичности) [7, 16, 20, 39].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 4)
Комментарии: Альтернативным методом лечения тяжелых фокальных дистоний и спастичности является Ботулинический токсин типа A-гемагглютинин комплекс**, Ботулинический токсин типа А**.
Терапия проводится аналогично таковой при детском церебральном параличе
- Рекомендуется терапия эпилептических приступов с индивидуальным подбором противоэпилептических препаратов пациентам с ГА1 с целью коррекции симптоматики [7, 16, 19, 20].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: Эпилептические приступы возникают у небольшого числа пациентов во время или после перенесенных энцефалитоподобных кризов, могут быть однократными и не повторяться в дальнейшем. В ряде случаев за эпилептические приступы врачи ошибочно принимают грубые гиперкинезы и дистонические атаки.
Сообщается, что монотерапия фенобарбиталом** и окскарбазепином** может купировать эпилептические приступы у пациентов этой категории [27, 28]. Однако, в большинстве случаев применяют политерапию такими противоэпилептическими препаратами, как леветирацетам**, ламотриджин, топирамат**, фенитоин**, карбамазепин** и клоназепам** [42-47].
- Не рекомендуется назначение препаратов солей вальпроевой кислоты** пациентам с ГА1 с целью предотвращения ухудшения состояния [2, 7, 16, 20, 21].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5)
Комментарии: вальпроаты выводятся из организма в виде вальпроилкарнитина, что истощает резерв свободного карнитина и угнетает работу дыхательной цепи митохондрий. Назначение препаратов вальпроевой кислоты**, таким образом, не желательны [21].
3.4.1 Лечение в период метаболического криза
Необходимо помнить, что острый энцефалитоподобный криз может развиться при любом инфекционном заболевании, вакцинации или хирургических вмешательствах в основном в возрасте до шести лет. Настораживающими симптомами, свидетельствующими о развитии декомпенсации по основному заболеванию, являются: повторные рвоты, диарея, неврологические расстройства (мышечная гипотония/ ригидность, повышенная возбудимость, мышечная дистония, угнетение сознания). У детей старше 6 лет риск развития острых неврологических нарушений во время кризов снижается, что связано с повышением толерантности мозга к колебаниям кислотно-щелочного состояния и воздействию токсических метаболитов.
Основными триггерами декомпенсации (факторами, вызывающие распад белка) являются
- Голодание
- Инфекционные процессы, особенно протекающие со рвотой, диареей
- Лихорадка (любой этиологии)
- Интенсивные физические упражнения
- Нарушение диеты (избыточное потребление белка)
Необходимо проведение тщательной клинической оценки состояния пациента, включая измерение артериального давления и оценку комы по шкале Глазго при подозрении на развитие метаболического криза.
Основными принципами лечения во время энцефалитоподобных кризов являются:
- восстановление катаболического статуса, путем назначения высокоэнергетической инфузионной терапии (декстроза** в сочетании с инсулинами и их аналогами);
- ограничение образования органических кислот, путем снижения поступления белка натуральных продуктов или полное их исключение (по возможности в течение 24 часов используют только специализированные продукты на основе смесей аминокислот)[28];
- введение высокодозного Левокарнитина;
- восстановление кислотно-щелочного состояния крови.
Подробные клинические и лабораторные показатели приведены в приложении А10.
- Рекомендуется коррекция патогенетической диетотерапии и доз левокарнитина пациентам с ГА1 в период метаболического криза с целью стабилизации состояния [7, 19, 20].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5)
Комментарии: Неотложная терапия должна начинаться немедленно и продолжаться весь период фебрильной лихорадки, после вакцинации, до и после хирургического вмешательства, особенно у пациентов в возрасте до 6 лет.
Назначают высокодозный #левокарнитин 150-200 мг/кг/сутки (приложение А10) [65].
#Левокарнитин вводится внутривенно –150-200 мг/кг/24 ч в виде 2 -4 раздельных доз или болюсно по 100 мг/кг в течение 30 минут с последующей медленной внутривенной инфузией по 4 мг/кг/ч. (максимальная доза 6 г/сут)[66].
Если внутривенное введение невозможно, возможен пероральный прием 200 мг/кг/сутки дробно.
- Рекомендуется инфузионная терапия в стационаре пациентам с ГА1 в период метаболического криза с целью предотвращения развития тяжелых неврологических нарушений [7, 19, 20].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5)
Комментарии: При появлении у больного рвоты, повышения температуры выше 38,50С, неврологических расстройств показана срочная госпитализация в стационар. Основные принципы интенсивной терапии в условиях стационара представлены в Приложении А10 [54]. Раннее начало интенсивного лечения позволяет предотвратить развитие тяжелых неврологических нарушений у большинства пациентов с ГА1. Внутривенная инфузионная терапия включает раствор декстрозы** из расчета 8-15 г/кг/сут, инсулинотерапию при гипергликемии (выше 10 ммоль/л) и/или глюкозурии. Не следует возмещать потери жидкости за счет энтерального питания (см. приложение А10) [28].
Инфузионная терапия включает:
- Внутривенное введение декстрозы** из расчета 8-15г/кг/сутки (Приложение А10). В случае гипогликемии - немедленное введение декстрозы** 200 мг/кг (2 мл/кг 10% декстрозы** или 1 мл/кг 20% декстрозы**) в течение нескольких минут.
- Внутривенное введение #Левокарнитина 150-200 мг/кг/сутки. Предпочтительно разводить Левокарнитин раствором декстрозы** или добавить в суточную инфузию декстрозы**.
В период метаболического криза необходим мониторинг клинических и лабораторных показателей (кислотно-щелочного состояния крови, глюкоза, электролиты (калий, натрий, кальций ионизированный, хлор) каждые 4-6 часов или раньше при наличии ухудшения или улучшений состояния пациентам с ГА1 в период метаболического криза с целью оценки состояния и своевременной коррекции терапии.
- Рекомендуется применение аминокислотных смесей без лизина и триптофана (или с низким содержанием триптофана) всем пациентам с ГА1 в период метаболического криза с целью восполнения потребности в белке за счет белкового эквивалента без патогенетически значимых аминокислот [28, 55].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5)
Комментарии: При невозможности перорального приема смеси рекомендовано кормление через назогастральный зонд. Применение специализированной смеси как единственного источника белка возможно в течение не более 48 часов, далее в питание необходимо введение источников натурального белка (грудное молоко, молочная смесь и др.). Расчет согласно приложению А9.
- Рекомендуется при рвоте применять противорвотные средства #Ондансетрон** в дозе 0,1- 0,15 мг/кг/сутки) в возрастных дозах с целью купирования симптома [55, 56, 57, 58].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5)
Комментарии: Приложение А10. Возможно применение других препаратов, обладающих противорвотным действием в возрастных дозировках.
- Рекомендуется при отечном синдроме дегидратационная терапия Фуросемидом** в возрастных дозах по клинической ситуации с целью нормализации водного баланса [55].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5)