Dasgupta A. Biotin and other interferences in immunoassays: a concise guide. Elsevier; 2019 Jan 15.
Leon‐Del‐Rio A. Biotin in metabolism, gene expression, and human disease. Journal of inherited metabolic disease. 2019 Jul;42(4):647-54.
Zempleni J, Hassan YI, Wijeratne SS. Biotin and biotinidase deficiency. Expert review of endocrinology & metabolism. 2008 Nov 1;3(6):715-24.
Hymes J, Stanley CM, Wolf B. Mutations in BTD causing biotinidase deficiency. Human mutation. 2001 Nov;18(5):375-81.
Deveci K, Akar HT, Yildiz Y, Özgül RK. A Novel Double Homozygous BTD Gene Mutation in A Case of Profound Biotinidase Deficiency. Türkiye Çocuk Hastalıkları Dergisi. 2022:1-3.
Küry S, Ramaekers V, Bézieau S, Wolf B. Clinical utility gene card for: biotinidase deficiency—update 2015. European Journal of Human Genetics. 2016 Jul;24(7):3-5.
Murry JB, Machini K, Ceyhan-Birsoy O, Kritzer A, Krier JB, Lebo MS, Fayer S, Genetti CA, VanNoy GE, Timothy WY, Agrawal PB. Reconciling newborn screening and a novel splice variant in BTD associated with partial biotinidase deficiency: a BabySeq Project case report. Molecular Case Studies. 2018 Aug 1;4(4):a002873.
Wiltink RC, Kruijshaar ME, van Minkelen R, Onkenhout W, Verheijen FW, Kemper EA, van Spronsen FJ, van der Ploeg AT, Niezen-Koning KE, Saris JJ, Williams M. Neonatal screening for profound biotinidase deficiency in the Netherlands: consequences and considerations. European Journal of Human Genetics. 2016 Oct;24(10):1424-9.
Procter M, Wolf B, Crockett DK, Mao R. The biotinidase gene variants registry: a paradigm public database. G3: Genes, Genomes, Genetics. 2013 Apr 1;3(4):727-31.
Norrgard KJ, Pomponio RJ, Swango KL, Hymes J, Reynolds T, Buck GA, Wolf B. Double mutation (A171T) and (D444H) is a common cause of profound biotinidase deficiency in children ascertained by newborn screening in the United States. Human mutation. 1998;11(5):410-.
Tammachote R, Janklat S, Tongkobpetch S, Suphapeetiporn K, Shotelersuk V. Holocarboxylase synthetase deficiency: novel clinical and molecular findings. Clinical genetics. 2010 Jul;78(1):88-93.
Suzuki Y, Yang X, Aoki Y, Kure S, Matsubara Y. Mutations in the holocarboxylase synthetase gene HLCS. Human Mutation. 2005 Oct;26(4):285-90.
Wang T, Ye J, Han LS, Qiu WJ, Zhang HW, Zhang YF, Gao XL, Wang Y, Gu XF. Diagnosis, treatment and gene mutation analysis in children with holocarboxylase synthetas deficiency. Zhongguo Dang dai er ke za zhi= Chinese Journal of Contemporary Pediatrics. 2009 Aug 1;11(8):609-12.
Wolf B. Biotinidase deficiency: new directions and practical concerns. Current treatment options in neurology. 2003 Jul;5(4):321-8.
Kuroishi T, Rios-Avila L, Pestinger V, Wijeratne SS, Zempleni J. Biotinylation is a natural, albeit rare, modification of human histones. Molecular genetics and metabolism. 2011 Dec 1;104(4):537-45.
Asgari A, Dehnabeh SR, Zargari M, Khani S, Mozafari H, Varasteh A, Keyfi F, Barzegari M, Hasanzaeh R, Khatami S. Clinical, biochemical and genetic analysis of biotinidase deficiency in Iranian population. Archives of Iranian Medicine. 2016 Nov 1;19(11):0-.
Canda E, Kalkan Uçar S, Çoker M. Biotinidase deficiency: prevalence, impact and management strategies. Pediatric health, medicine and therapeutics. 2020 May 4:127-33.
Wu HR, Chen KJ, Hsiao HP, Chao MC. Impaired glucose homeostasis and a novel HLCS pathogenic variant in holocarboxylase synthetase deficiency: a report of two cases and brief review. Journal of Pediatric Endocrinology and Metabolism. 2020 Nov 1;33(11):1481-6.
Зыков ВП, Заваденко АН, Милованова ОА, Степанищев ИЛ, Самигулина МГ. Недостаточность биотинидазы. Медицинский совет. 2009(1):39-44.
Afroze B, Wasay M. Biotinidase deficiency in Pakistani children; what needs to be known and done.
Salbert BA, Pellock JM, Wolf B. Characterization of seizures associated with biotinidase deficiency. Neurology. 1993 Jul 1;43(7):1351-.
Wolf B, Heard GS, Weissbecker KA, McVoy JR, Grier RE, Leshner RT. Biotinidase deficiency: initial clinical features and rapid diagnosis. Annals of neurology. 1985 Nov;18(5):614-7.
Baumgartner MR, Suormala T. Biotin-responsive disorders. Inborn metabolic diseases: diagnosis and treatment. 2016:375-83.
Taitz LS, Leonard JV, Bartlett K. Long-term auditory and visual complications of biotinidase deficiency. Early human development. 1985 Sep 1;11(3-4):325-31.
Weber P, Scholl S, Baumgartner ER. Outcome in patients with profound biotinidase deficiency: relevance of newborn screening. Developmental medicine and child neurology. 2004 Jul;46(7):481-4.
Salbert BA, Astruc J, Wolf B. Ophthalmologic findings in biotinidase deficiency. Ophthalmologica. 1993;206(4):177–181.
Burton BK, Roach ES, Wolf B, Weisbecker KA. Sudden death associated with biotinidase deficiency. Pediatrics. 1987 Mar;79(3):482-3.
Wolf B. Clinical issues and frequent questions about biotinidase deficiency. Molecular genetics and metabolism. 2010 May 1;100(1):6-13.
Ferreira P, Chan A, Wolf B. Irreversibility of symptoms with biotin therapy in an adult with profound biotinidase deficiency. JIMD Reports, Volume 36. 2017:117-20.
Ramaekers VT, Suormala TM, Brab M, Duran R, Heimann G, Baumgartner ER. A biotinidase Km variant causing late onset bilateral optic neuropathy. Archives of disease in childhood. 1992 Jan 1;67(1):115-9.
Wolf B. The neurology of biotinidase deficiency. Molecular genetics and metabolism. 2011 Sep 1;104(1-2):27-34.
Lott IT, Lottenberg S, Nyhan WL, Buchsbaum MJ. Cerebral metabolic change after treatment in biotinidase deficiency. Journal of inherited metabolic disease. 1993 Mar; 16:399-407.
Wolf B, Grier RE, Secor McVoy JR, Heard GS. Biotinidase deficiency: a novel vitamin recycling defect. Journal of inherited metabolic disease. 1985 Mar; 8:53-8.
Suchy SF, Rizzo WB, Wolf B. Fatty acids in biotin deficiency. Annals of the New York Academy of Sciences. 1985 Jun;447(1):429-.
Sander JE, Malamud N, Cowan MJ, Packman S, Amman AJ, Wara DW. Intermittent ataxia and immunodeficiency with multiple carboxylase deficiencies: a biotin‐responsive disorder. Annals of Neurology: Official Journal of the American Neurological Association and the Child Neurology Society. 1980 Nov;8(5):544-7.
Regula Baumgartner E, Suormala TM, Wick H, Probst A, Blauenstein U, Bachmann C, Vest M. Biotinidase deficiency: a cause of subacute necrotizing encephalomyelopathy (Leigh syndrome). Report of a case with lethal outcome. Pediatric research. 1989 Sep;26(3):260-6.
Chedrawi AK, Ali A, Al Hassnan ZN, Faiyaz-Ul-Haque M, Wolf B. Profound biotinidase deficiency in a child with predominantly spinal cord disease. Journal of child neurology. 2008 Sep;23(9):1043-8.
Wiznitzer M, Bangert BA. Biotinidase deficiency: clinical and MRI findings consistent with myelopathy. Pediatric neurology. 2003 Jul 1;29(1):56-8.
Wolf B, Spencer R, Gleason T. Hearing loss is a common feature of symptomatic children with profound biotinidase deficiency. The Journal of pediatrics. 2002 Feb 1;140(2):242-6.
Porta F, Pagliardini V, Celestino I, Pavanello E, Pagliardini S, Guardamagna O, Ponzone A, Spada M. Neonatal screening for biotinidase deficiency: A 30-year single center experience. Molecular genetics and metabolism reports. 2017 Dec 1;13:80-2.
Baykal T, Gokcay G, Gokdemir Y, Demir F, Seckin Y, Demirkol M, Jensen K, Wolf B. Asymptomatic adults and older siblings with biotinidase deficiency ascertained by family studies of index cases. Journal of Inherited Metabolic Disease: Official Journal of the Society for the Study of Inborn Errors of Metabolism. 2005 Dec;28(6):903-12.
Wolf B. Successful outcomes of older adolescents and adults with profound biotinidase deficiency identified by newborn screening. Genetics in Medicine. 2017 Apr;19(4):396-402.
Wolf B. Worldwide survey of neonatal screening for biotinidase deficiency. Journal of inherited metabolic disease. 1991 Nov;14(6):923-7.
Sivri HS, Genç GA, Tokatlı A, Dursun A, Coşkun T, Aydın Hİ, Sennaroğlu L, Belgin E, Jensen K, Wolf B. Hearing loss in biotinidase deficiency: genotype-phenotype correlation. The Journal of pediatrics. 2007 Apr 1;150(4):439-42.
Wolf B. Biotinidase deficiency:“if you have to have an inherited metabolic disease, this is the one to have”. Genetics in Medicine. 2012 Jun 1;14(6):565-75
Donti TR, Blackburn PR, Atwal PS. Holocarboxylase synthetase deficiency pre and post newborn screening. Molecular genetics and metabolism reports. 2016 Jun 1; 7:40-4.
Wu HR, Chen KJ, Hsiao HP, Chao MC. Impaired glucose homeostasis and a novel HLCS pathogenic variant in holocarboxylase synthetase deficiency: a report of two cases and brief review. Journal of Pediatric Endocrinology and Metabolism. 2020 Nov 1;33(11):1481-6.
Søvik O. Inborn errors of amino acid and fatty acid metabolism with hypoglycemia as a major clinical manifestation. Acta Pædiatrica. 1989 Mar;78(2):161-70.
Bandaralage SP, Farnaghi S, Dulhunty JM, Kothari A. Antenatal and postnatal radiologic diagnosis of holocarboxylase synthetase deficiency: a systematic review. Pediatric radiology. 2016 Mar; 46:357-64.
Saleem H, Simpson B. Biotinidase Deficiency. InStatPearls [Internet] 2022 Nov 27. StatPearls Publishing.
Bilge N, Yevgi R. Biotinidase deficiency in differential diagnosis of neuromyelitis optica spectrum disorder. Multiple Sclerosis and Related Disorders. 2020;44, 102280.
Горошко ЛВ. Клиническое наблюдение недостаточности биотинидазы. Российский вестник перинатологии и педиатрии. 2013;58(1):29-30.
Heard GS, Wolf B, Jefferson LG, Weissbecker KA, Nance WE, McVory JR, Napolitano A, Mitchell PL, Lambert FW, Linyear AS. Neonatal screening for biotinidase deficiency: results of a 1-year pilot study. The Journal of pediatrics. 1986 Jan 1;108(1):40-6.
Cowan TM, Blitzer MG, Wolf B, of the American AW. Technical standards and guidelines for the diagnosis of biotinidase deficiency. Genetics in Medicine. 2010 Jul 1;12(7):464-70.
Cicalini I, Pieragostino D, Rizzo C, Verrocchio S, Semeraro D, Zucchelli M, Di Michele S, Dionisi-Vici C, Stuppia L, De Laurenzi V, Bucci I. Partial Biotinidase Deficiency Revealed Imbalances in Acylcarnitines Profile at Tandem Mass Spectrometry Newborn Screening. International Journal of Environmental Research and Public Health. 2021 Feb;18(4):1659.
Tankeu, A. T., Van Winckel, G., Elmers, J., Jaccard, E., Superti-Furga, A., Wolf, B., & Tran, C. (2023). Biotinidase deficiency: What have we learned in forty years? Molecular Genetics and Metabolism, 138(4), 107560.
Swango KL, Demirkol M, Hüner G, Pronicka E, Sykut-Cegielska J, Schulze A, Wolf B. Partial biotinidase deficiency is usually due to the D444H mutation in the biotinidase gene. Human genetics. 1998 May; 102:571-5.
Manual IC. Inborn errors of metabolism. UCSF Children’s Hospital at UCSF Medical Center. 2016.
Altun I, Kiykim A, Zubarioglu T, Burtecene N, Hopurcuoglu D, Topcu B, Cansever MS, Kiykim E, Cokugras HC, Aktuglu Zeybek AC. Altered immune response in organic acidemia. Pediatrics International. 2022 Jan;64(1):e15082.
Baker PR. Pathophysiology of Inherited Metabolic Diseases. In Nutrition Management of Inherited Metabolic Diseases: Lessons from Metabolic University 2022 Jun 15 (pp. 33-43). Cham: Springer International Publishing.
Prietsch V, Lindner M, Zschocke J, Nyhan WL, Hoffmann GF. Emergency management of inherited metabolic diseases. Journal of inherited metabolic disease. 2002 Nov;25(7):531-46.
del Pilar Chantada-Vázquez M, Bravo SB, Barbosa-Gouveia S, Alvarez JV, Couce ML. Proteomics in Inherited Metabolic Disorders. International Journal of Molecular Sciences. 2022 Dec 1;23(23):14744.
Reddy N, Calloni SF, Vernon HJ, Boltshauser E, Huisman TA, Soares BP. Neuroimaging findings of organic acidemias and aminoacidopathies. Radiographics. 2018 May;38(3):912-31.
Wolf B. Biotinidase Deficiency Synonym: Late-Onset Multiple Carboxylase Deficiency. Gene Reviews: https://www. ncbi. nlm. nih. gov/books/NBK1322. 2016.
Reynolds E, Blanchard S, Jalazo E, Chakraborty P, Bailey Jr DB. Newborn Screening Conditions: Early Intervention and Probability of Developmental Delay. Journal of Developmental & Behavioral Pediatrics. 2022 May 13:10-97.
Alliance G. Understanding genetics: a district of Columbia guide for patients and health professionals.
VanVleck N, Wolf B, Seeterlin M, Monaghan KG, Stanley E, Hawkins H, Taffe B. Improved Identification of Partial Biotinidase Deficiency by Newborn Screening Using Age-Related Enzyme Activity Cutoffs: Reduction of the False-Positive Rate. International Journal of Neonatal Screening. 2015 Jun 19;1(1):45-56.
British National Formularyfor Children; British Medical Association and Royal Pharmaceutical Society of Great Britain. London.
Галиева Галина Юрьевна, Федосеева Ирина Фаисовна, Бедарева Татьяна Юрьевна, & Урбан Елена Николаевна (2019). Клинический случай недостаточности биотинидазы у ребенка раннего возраста. Мать и дитя в Кузбассе, (4), 75-78.
György P, Rose CS, Eakin RE, Snell EE, Williams RJ. Egg-white injury as the result of nonabsorption or inactivation of biotin. Science. 1941 May 16;93(2420):477-8.
Анисимова И.В. и др. Методические рекомендации «Метод получения сухого пятна крови на тест-бланк для проведения клинико-лабораторных исследований». 2022:34.
Canda E, Kalkan Uçar S, Çoker M. Biotinidase Deficiency: Prevalence, Impact And Management Strategies. Pediatric Health Med Ther. 2020; 11:127-133. Published 2020 May 4. doi:10.2147/PHMT.S198656
Worthen HG, al Ashwal A, Ozand PT, et al. Comparative frequency and severity of hypoglycemia in selected organic acidemias, branched chain amino acidemia, and disorders of fructose metabolism. Brain Dev. 1994;16 Suppl:81-85. doi:10.1016/0387-7604(94)90100-7
Wolf B. Biotinidase Deficiency. 2000 Mar 24 [Updated 2023 May 25]. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1322/
Devanapalli B, Sze Hui Wong R, Lim N, Ian Andrews P, Vijayan K, Kim WT, et al.Biotinidase deficiency: A treatable neurometabolic disorder. Brain Dev CaseRep. 2024;2(2):100021.
Demirtürk Z, Şentürk E, Köse A, Özcan PE, Telci L. A Case of Biotinidase Deficiency in an Adult with Respiratory Failure in the Intensive Care Unit. Balkan Med J. 2016;33(5):563-565. doi:10.5152/balkanmedj.2016.150359
Santoro JD, Paulsen KC. Biotinidase Deficiency as a Mimic of Neuromyelitis Optica Spectrum Disorder in Childhood. JAMA Neurol. Published online October 5, 2020. doi:10.1001/jamaneurol.2020.3558
Rajesh C, Babji NS, Siddiq MAM. Case report of early biotinidase deficiency, a type of multiple carboxylase deficiency. Int J Contemp Pediatr 2021;8:1290-2
Неонатальный скрининг: национальное руководство / под ред. С.И. Куцева. Москва: ГЭОТАР-Медиа, 2023. — 360 с. — (Серия «Национальные руководства»). — DOI: 10.33029/9704-7737-3-NEO-2023-1-360.
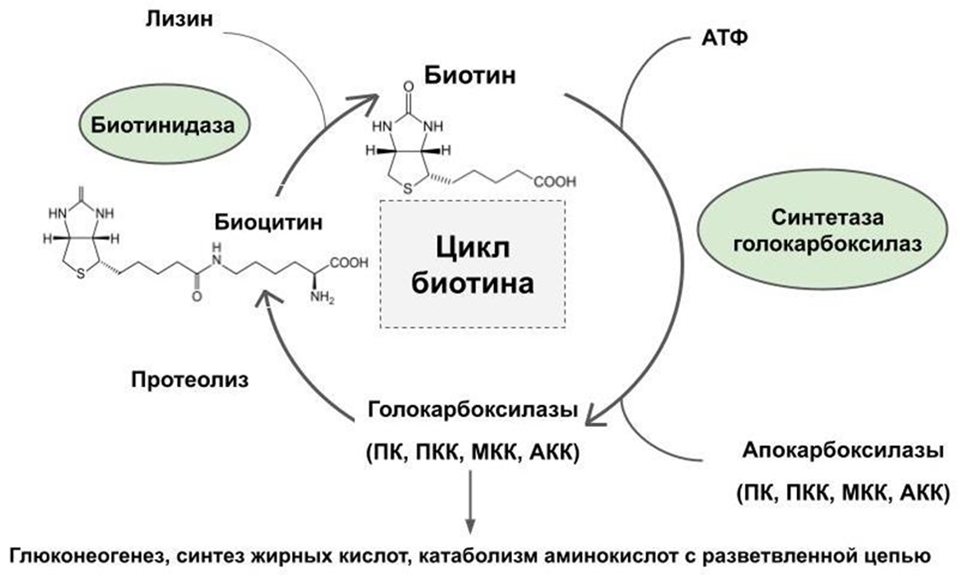 Рисунок 1. Цикл биотина.
Рисунок 1. Цикл биотина.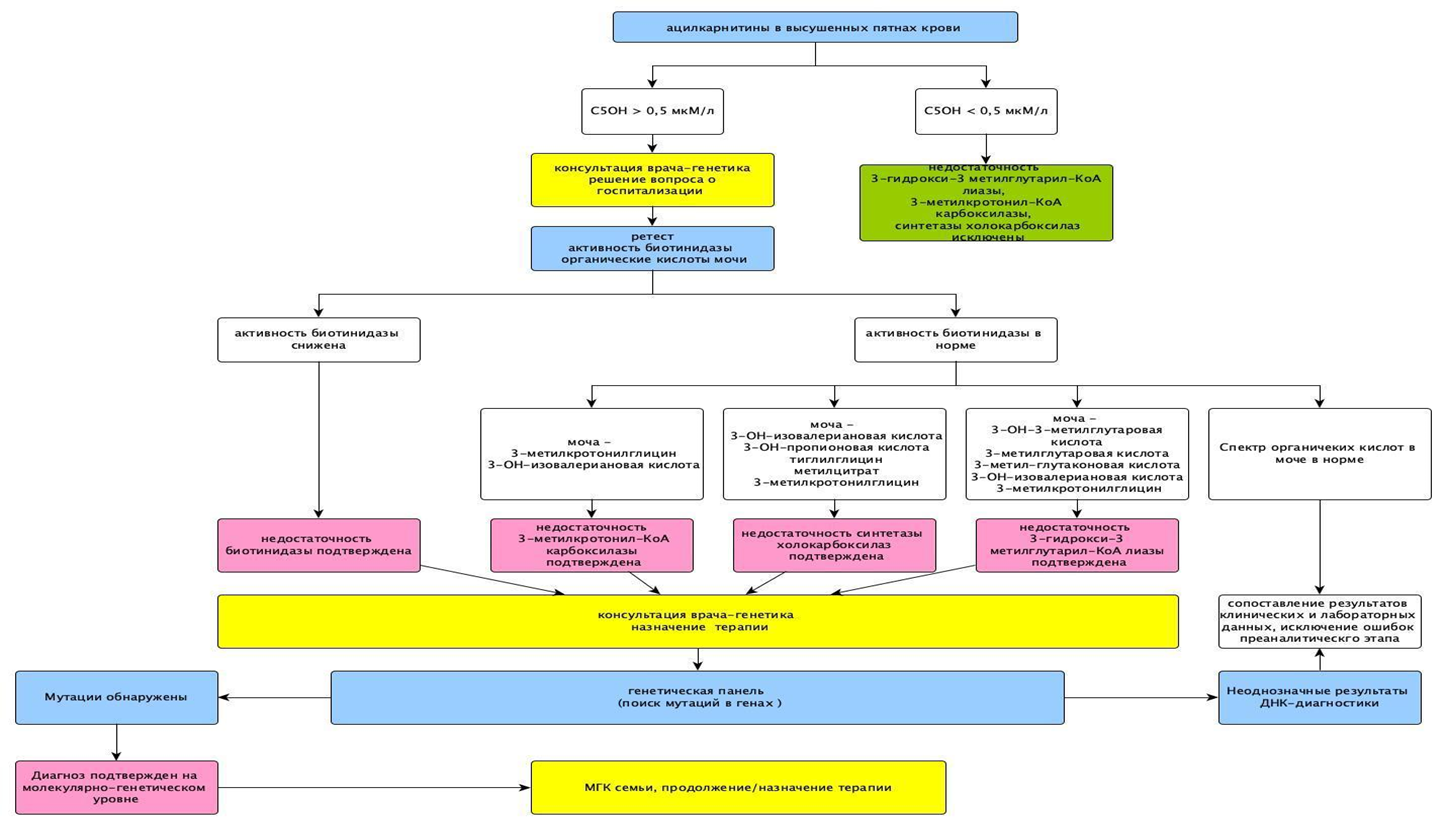 Рисунок 1Б. Алгоритм неонатального скрининга на выявление недостаточности биотинидазы (НБ) и недостаточности синтетазы холокарбоксилаз (голокарбоксилаз) (НСГ) и других заболеваний, ассоциированных с изменением уровня 3-гидроксиизовалерилкарнитина (C5OH): недостаточность 3-метилкротонил-КоА карбоксилазы и недостаточность 3-ОН-3 метилглутарил КоА лиазы.
Рисунок 1Б. Алгоритм неонатального скрининга на выявление недостаточности биотинидазы (НБ) и недостаточности синтетазы холокарбоксилаз (голокарбоксилаз) (НСГ) и других заболеваний, ассоциированных с изменением уровня 3-гидроксиизовалерилкарнитина (C5OH): недостаточность 3-метилкротонил-КоА карбоксилазы и недостаточность 3-ОН-3 метилглутарил КоА лиазы.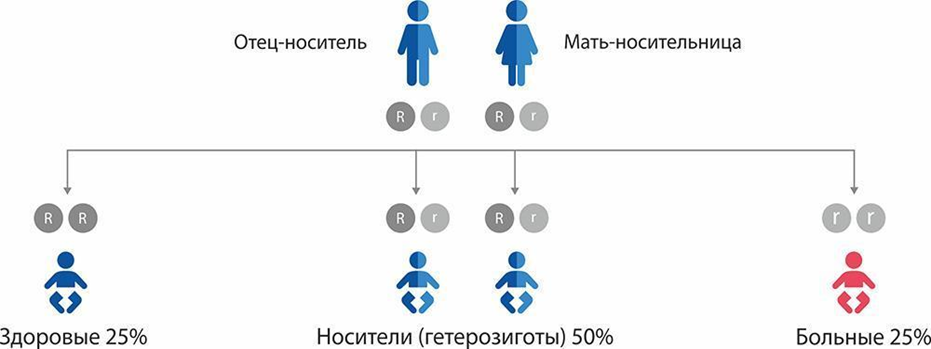 Рисунок 1В. Схема наследования недостаточности биотинидазы (НБ) и недостаточности синтетазы голокарбоксилаз (НСГ). При наличии двух копий «больного» гена («r»), унаследованных от обоих родителей-носителей, риск рождения ребенка с НБ и НСГ составляет 25% («rr»).
Рисунок 1В. Схема наследования недостаточности биотинидазы (НБ) и недостаточности синтетазы голокарбоксилаз (НСГ). При наличии двух копий «больного» гена («r»), унаследованных от обоих родителей-носителей, риск рождения ребенка с НБ и НСГ составляет 25% («rr»).


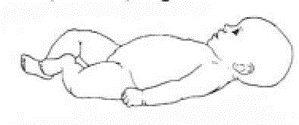

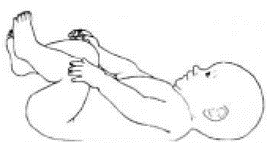
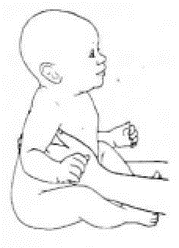
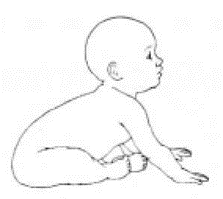
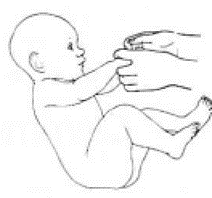
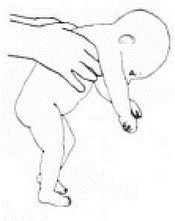
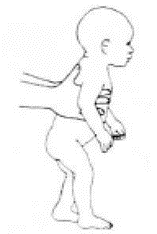
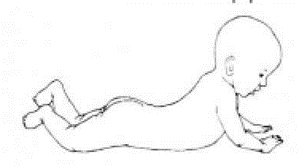






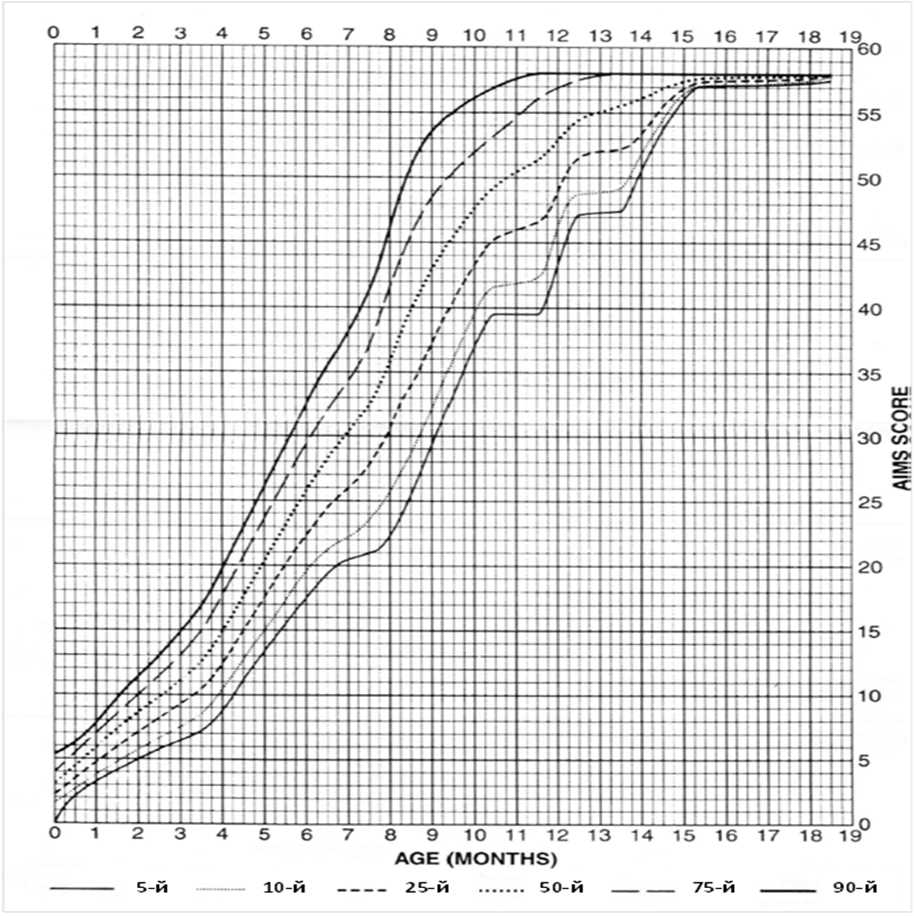 Приложение Г2. Забор биоматериала для диагностики в пятнах крови
Приложение Г2. Забор биоматериала для диагностики в пятнах крови