Orphanet website (дата посещения 28.03.2021) URL:https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=134
Sturm E., Piersma F.E., Tanner M.S. et al. Controversies and Variation in Diagnosing and Treating Children With Wilson Disease: Results of an International Survey. J Pediatr Gastroenterol Nutr. 2016; 63 (1): 82-7.
European Association for Study of Liver. EASL Clinical Practice Guidelines: Wilson"s disease. J Hepatol. 2012; 56 (3): 671-85.
Roberts E.A., Schilsky M.L. Diagnosis and treatment of Wilson disease: an update. Hepatology. 2008; 47 (6): 2089-111.
Четкина Т.С., Потапов А.С., Цирульникова О.М. и др. Болезнь Вильсона у детей: варианты манифестации и трудности ранней диагностики. Вопросы диагностики в педиатрии. 2011; 3 (1): 41-47.
Четкина Т.С. Болезнь Вильсона у детей. Автореф. дисс. … канд. мед. Наук. 2011. 24 с.
Голованова Е.В., Лазебник Л.Б., Конев Ю.В. и др. Болезнь Вильсона-Коновалова (гепатоцеребральная дистрофия, гепатолентикулярная дегенерация): диагностика, лечение и диспансерное наблюдение. Экспериментальная и клиническая гастроэнтерология. 2015; 7 (119): 108-111.
Соколов А.А., Дембровский В.Н., Красильникова Е.Ю. Оказание медицинской помощи и лекарственное обеспечения пациентов, страдающих жизнеугрожающими и хроническими прогрессирующими редкими заболеваниями. Болезнь Вильсона (гепатолентикулярная дегенерация). Проблемы стандартизации в здравоохранении. 2015; 5-6: 30-35.
Корой П.В. Болезнь Вильсона. Часть I: этиология, патогенез, клинические проявления и скрининг. Вестник молодого ученого. 2014; 7 (3-4): 56-63.
Корой П.В. Болезнь Вильсона. Часть II: диагностические тесты, диагностика в специфических популяциях. Вестник молодого ученого. 2015; 8 (1): 21-30.
Корой П.В. Болезнь Вильсона. Часть III: общие принципы терапии, лечение в специальных ситуациях, мониторинг терапии и прогноз. Вестник молодого ученого. 2015; 9 (2): 35-44.
Saroli Palumbo C, Schilsky M.L. Clinical practice guidelines in Wilson disease. Ann Transl Med. 2019 Apr;7(Suppl 2):S65.
Kumar M., Gaharwar U., Paul S. et al. WilsonGen a comprehensive clinically annotated genomic variant resource for Wilson"s Disease. Sci Rep. 2020 Jun 3;10(1):9037.
Балашова М.С., Соловьева О.В., Фастовец С.В. и др. Клиническая ценность секвенирования гена ATP7B в диагностике болезни Вильсона-Коновалова. Медицинская генетика. 2016; 15 (7): 14-16.
Ferenci P. Regional distribution of mutations of the ATP7B gene in patients with Wilson disease: impact on genetic testing. Hum Genet. 2006 Sep;120(2):151-9.
Гернер Е.А., Назаров В.Д., Федорова Т.Ф. и др. Клинико-лабораторная и молекулярно-генетическая диагностика болезни Вильсона-Коновалова. Российский неврологический журнал. 2019;24(3):10–18
Баязутдинова Г.М., Щагина О.А., Поляков А.В. Мутация с.3207C>A гена АТР7В – наиболее частая причина гепатолентикулярной дегенерации в России: частота и причина распространения. Медицинская генетика. 2018;17(4):25-30.
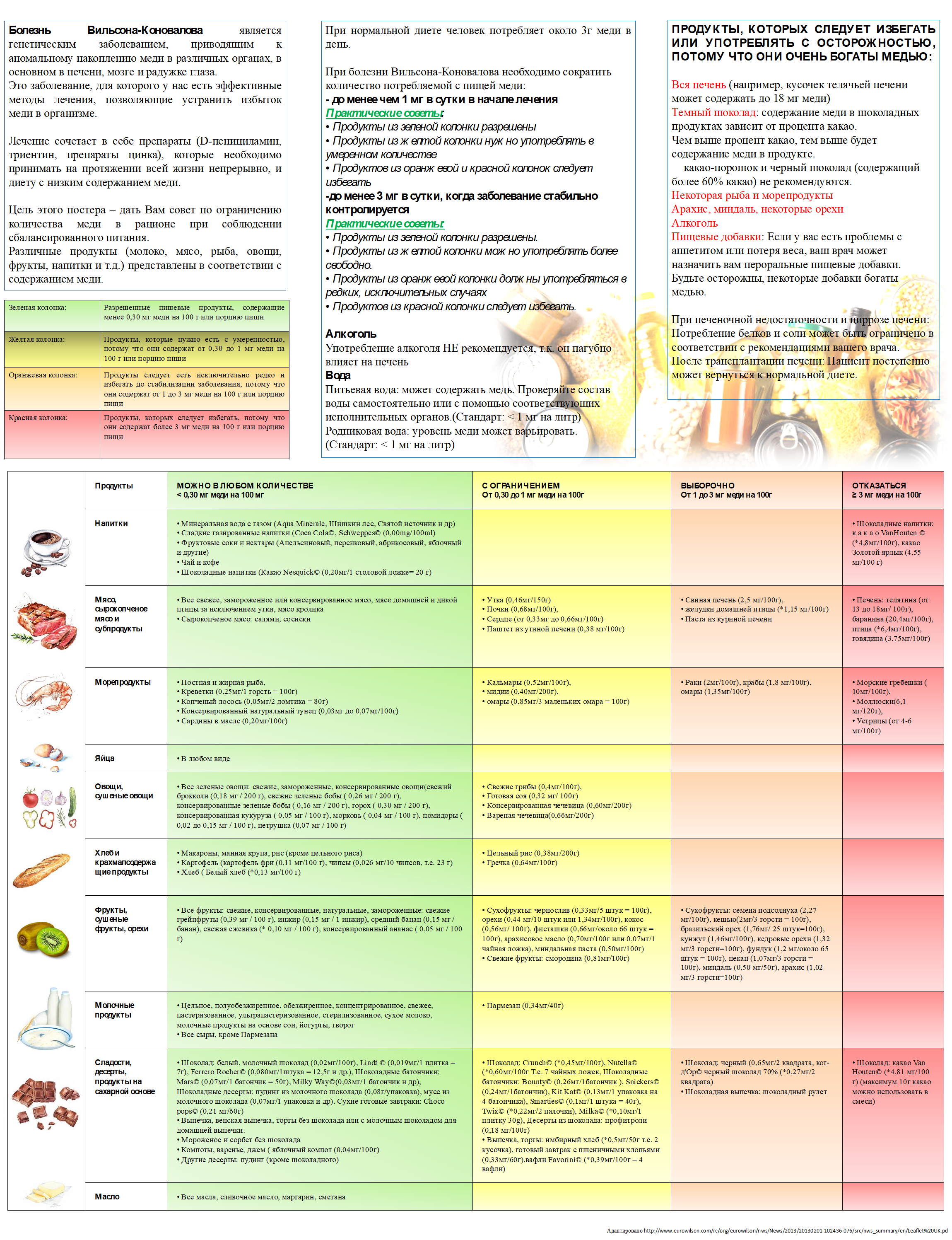
Нормы физиологических потребностей в энергии и пищевых веществах для различных групп населения Российской Федерации. Методические рекомендации МР 2.3.1.2432—08
Scheiber IF, Brůha R, Dušek P. Pathogenesis of Wilson disease. Handb Clin Neurol. 2017;142:43-55.
Coffey A.J., Durkie M., Hague S. et al. A genetic study of Wilson"s disease in the United Kingdom. Brain. 2013 May;136(Pt 5):1476-87.
Bandmann O., Weiss K.H., Kaler S.G. Wilson"s disease and other neurological copper disorders. Lancet Neurol. 2015 Jan;14(1):103-13.
Scheinberg IH, Sternlieb I. Wilson"s disease. In: Smith LH, ed. Major problems in internal medicine. Vol 23. Philadelphia: WB Saunders, 1984.
Park R.H., McCabe P., Fell G.S. et al. Wilson"s disease in Scotland. Gut. 1991 Dec;32(12):1541-5.
Hahn S.H., Lee S.Y., Jang Y.J. et al. Pilot study of mass screening for Wilson"s disease in Korea. Mol Genet Metab. 2002 Jun;76(2):133-6.
Красильникова Е.Ю., Соколов А.А. Анализ ситуации в сфере оказания медицинской помощи и лекарственного обеспечения пациентов, страдающих редкими заболеваниями, в период 2013-2015 годов. Проблемы стандартизации в здравоохранении. 2016; 3-4
Ferenci P., Caca K., Loudianos G. et al. Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003 Jun;23(3):139-42.
Коновалов Н.В. Гепатолентикулярная дистрофия. M., 1960. 556 с.
Hermann W. Classification and differential diagnosis of Wilson"s disease. Ann Transl Med. 2019 Apr;7(Suppl 2):S63.
Lin L.J.,Wang D.X., Ding N.N. et al. Comprehensive analysis on clinical features of Wilson’s disease: an experience over 28 years with 133 cases. Neurol Res 2014;36:157–63.
Wilson D.C., Phillips M.J., Cox D.W. et al. Severe hepatic Wilson"s disease in preschool-aged children. J Pediatr. 2000 Nov;137(5):719-22.
Socha P., Janczyk W., Dhawan A. et al. Wilson"s Disease in Children: A Position Paper by the Hepatology Committee of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr. 2018 Feb;66(2):334-344.
Machado A, Chien HF, Deguti MM, et al. Neurological manifestations in Wilson"s disease: Report of 119 cases. Mov Disord. 2006 Dec;21(12):2192-6.
Abdel Ghaffar TY, Elsayed SM, Elnaghy S, et al. Phenotypic and genetic characterization of a cohort of pediatric Wilson disease patients. BMC Pediatr. 2011 Jun 17;11:56.
Lorincz M.T. Neurologic Wilson"s disease. Ann N Y Acad Sci. 2010 Jan;1184:173-87.
Azizi E, Eshel G, Aladjem M. Hypercalciuria and nephrolithiasis as a presenting sign in Wilson disease. Eur J Pediatr 1989;148:548-549.
Chu CC, Huang CC, Chu NS. Recurrent hypokalemic muscle weakness as an initial manifestation of Wilson’s disease. Nephron 1996;73:477-479.
Golding D.N., Walshe J.M. Arthropathy of Wilson’s disease. Study of clinical and radiological features in 32 patients. Ann Rheum Dis 1977; 36:99-111.
Hlubocka Z., Maracek Z., Linhart A. et al. Cardiac involvement in Wilson disease. J Inherit Metab Dis 2002;25:269-277.
Sanchez-Albisua I., Garde T., Hierro L. et al. A high index of suspicion: the key to an early diagnosis of Wilson’s disease in childhood. J Pediatr Gastroenterol Nutr 1999;28:186–190.
Merle U, Schaefer M, Ferenci P, et al. Clinical presentation, diagnosis and long-term outcome of Wilson’s disease: a cohort study. Gut 2007;56:115–20.
Tauber J, Steinert RF. Pseudo-Kayser-Fleischer ring of the cornea associated with non-Wilsonian liver disease. A case report and literature review. Cornea 1993;12:74-77.
Dunn LL, Annable WL, Kliegman RM. Pigmented corneal rings in neonates with liver disease. J Pediatr 1987;110:771-776
Walshe JM. The acute haemolytic syndrome in Wilson’s disease—a review of 22 patients. QJM 2013;106:1003–8.
Pfeiffer RF. Wilson Disease. Continuum (Minneap Minn). 2016 Aug;22(4 Movement Disorders):1246-61.
Schilsky ML. Wilson Disease: Diagnosis, Treatment, and Follow-up. Clin Liver Dis. 2017 Nov;21(4):755-767.
Sallie R, Katsiyiannakis L, Baldwin D, et al. Failure of simple biochemical indexes to reliably differentiate fulminant Wilson’s disease from other causes of fulminant liver failure. Hepatology 1992; 16:1206–11.
Nazer H, Ede RJ, Mowat AP, Williams R. Wilson’s disease: clinical presentation and use of prognostic index. Gut 1986;27:1377-1381.
Ryan A, Nevitt SJ, Tuohy O, Cook P. Biomarkers for diagnosis of Wilson"s disease. Cochrane Database Syst Rev. 2019 Nov 19;2019(11):CD012267.
Gong A, Leitold S, Uhanova J, Minuk GY. Non-Wilson"s Disease-Associated Hypoceruloplasminemia. J Clin Exp Hepatol. 2020 Jul-Aug;10(4):284-289
Gromadzka G, Schmidt HH, Genschel J, et al. Frameshift and nonsense mutations in the gene for ATPase7B are associated with severe impairment of copper metabolism and with an early clinical manifestation of Wilson’s disease. Clin Genet 2005;68:524–32.
Merle U, Eisenbach C, Weiss KH, et al. Serum ceruloplasmin oxidase activity is a sensitive and highly specific diagnostic marker for Wilson’s disease. J Hepatol 2009;51:925–30.
Kelly J, Raizman JE, Bevilacqua V, et al. Complex reference value distributions and partitioned reference intervals across the pediatric age range for 14 specialized biochemical markers in the CALIPER cohort of healthy community children and adolescents. Clin Chim Acta 2015;450:196–202.
Giacchino R, Marazzi MG, Barabino A. et al. Syndromic variability of Wilson"s disease in children. Clinical study of 44 cases. Ital J Gastroenterol Hepatol. 1997 Apr;29(2):155-61.
Nicastro E, Ranucci G, Vajro P, et al. Re-evaluation of the diagnostic criteria for Wilson disease in children with mild liver disease. Hepatology 2010;52:1948–56.
Müller T, Koppikar S, Taylor RM. et al. Re-evaluation of the penicillamine challenge test in the diagnosis of Wilson"s disease in children. J Hepatol. 2007 Aug;47(2):270-6.
Ивлева С.А., Дворяковская Г.М., Четкина Т.С. и др. Диагностика фиброза печени у детей с болезнью Вильсона. Российский педиатрический журнал. 2014; 17 (3): 9-16.
Poujois A, Woimant F. Wilson"s disease: A 2017 update. Clin Res Hepatol Gastroenterol. 2018 Dec;42(6):512-520.
Paternostro R., Pfeiffenberger J., Ferenci P. et al. Non-invasive diagnosis of cirrhosis and long-term disease monitoring by transient elastography in patients with Wilson disease. Liver International. 2020;40:894–904.
Karlas T., Hempel M., Tröltzsch M. et al. Non-invasive evaluation of hepatic manifestation in Wilson disease with transient elastography, ARFI, and different fibrosis scores. Scand J Gastroenterol. 2012 Nov;47(11):1353-61
van Wassenaer-van Hall HN, van den Heuvel AG, Algra A. et al. Wilson disease: findings at MR imaging and CT of the brain with clinical correlation. Radiology. 1996 Feb;198(2):531-6.
Członkowska A, Litwin T, Chabik G. Wilson disease: neurologic features. Handb Clin Neurol. 2017;142:101-119.
Pandey N, John S. Kayser-Fleischer Ring. 2020 Aug 11. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021 Jan–. PMID: 29083643.
Ferenci P, Steindl-Munda P, Vogel W. et al. Diagnostic value of quantitative hepatic copper determination in patients with Wilson"s Disease. Clin Gastroenterol Hepatol. 2005 Aug;3(8):811-8.
Liggi M, Mais C, Demurtas M. et al. Uneven distribution of hepatic copper concentration and diagnostic value of double-sample biopsy in Wilson"s disease. Scand J Gastroenterol. 2013 Dec;48(12):1452-8.
Yang X, Tang XP, Zhang YH. Et al. Prospective evaluation of the diagnostic accuracy of hepatic copper content, as determined using the entire core of a liver biopsy sample. Hepatology. 2015 Dec;62(6):1731-41.
Johncilla M, Mitchell KA. Pathology of the liver in copper overload. Semin Liver Dis. 2011 Aug;31(3):239-44.
Волынец Г.В., Евлюхина Н.Н., Потапов А.С. и др. Взаимосвязь степени нарушения функции печени и ее морфологических изменений при болезни Вильсона у детей. Экспериментальная и клиническая гастроэнтерология. 2015; 5 (117): 82.
Сурков А.Н., Потапов А.С., Туманова Е.Л. и др. Гистопатологические изменения печени при болезни Вильсона у детей. Российский журнал гастроэнтерологии, гепатологии, колопроктологии. 2009; 19 (1) s33: 69.
Lau J.Y., Lai C.L., Wu P.C. et al. Wilson’s disease: 35 years experience. Q J Med. 1990 Jun;75(278):597-605.
Czlonkowska A., Gajda J., Rodo M. et al. Effects of long-term treatment in Wilson’s disease with D-penicillamine and zinc sulphate. J Neurol. 1996 Mar;243(3):269-73.
Weiss K.H., Thurik F., Gotthardt D.N. et al. Efficacy and safety of oral chelators in treatment of patients with Wilson disease. Clin Gastroenterol Hepatol. 2013 Aug;11(8):1028-35.e1-2.
Czlonkowska A., Litwin T., Dusek P. et al. Nature Reviews disease primers article: Wilson disease. 2019 Mar; Nat Rev Dis Primers.;4(1):21.
Yuzbasiyan-Gurkan V., Grider A., Nostrant T. et al. Treatment of Wilson’s disease with zinc: X. Intestinal metallothionein induction. J Lab Clin Med. 1992 Sep;120(3):380-6.
Sturniolo G.C., Mestriner C., Irato P. et al. Zinc therapy increases duodenal concentrations of metallothionein and iron in Wilson’s disease patients. Am J Gastroenterol. 1999 Feb;94(2):334-8.
Brewer GJ, Dick RD, Johnson VD et al. Treatment of Wilson’s disease with zinc: XV Long-term follow-up studies. J Lab Clin Med 1998;132:264-278.
Eda K, Mizuochi T, Iwama I, Inui A, Etani Y, Araki M, Hara S, Kumagai H, Hagiwara SI, Murayama K, Murakami J, Shimizu N, Kodama H, Yasuda R, Takaki Y, Yamashita Y. Zinc monotherapy for young children with presymptomatic Wilson disease: A multicenter study in Japan. J Gastroenterol Hepatol. 2018 Jan;33(1):264-269.
Brewer GJ, Dick RD, Johnson VD et al. Treatment of Wilson’s disease with zinc XVI: Treatment during the pediatric years. J Lab Clin Med 2001;137:191-198.
Wiernicka A, Jańczyk W, Dądalski M, Avsar Y, Schmidt H, Socha P. Gastrointestinal side effects in children with Wilson"s disease treated with zinc sulphate. World J Gastroenterol. 2013 Jul 21;19(27):4356-62.
Mizuochi T, Kimura A, Shimizu N, et al. Zinc monotherapy from time of diagnosis for young pediatric patients with presymptomatic Wilson disease. J Pediatr Gastroenterol Nutr 2011;53:365–7.
Ranucci G, Di Dato F, Spagnuolo MI, et al. Zinc monotherapy is effective inWilson’s disease patients with mild liver disease diagnosed in childhood: a retrospective study. Orphanet J Rare Dis 2014;9:41.
Abuduxikuer K, Wang JS. Zinc mono-therapy in pre-symptomatic Chinese children with Wilson disease: a single center, retrospective study. PLoS One 2014;9:e86168.
Marcellini M, Di Ciommo V, Callea F, et al. Treatment of Wilson’s disease with zinc from the time of diagnosis in pediatric patients: a single-hospital, 10-year follow-up study. J Lab Clin Med 2005;145:139–43.
Ahmad, A., Torrazza-Perez, E. & Schilsky, M. L. Liver transplantation for Wilson disease. Handb Clin. Neurol. 142, 193–204 (2017).
Michael L Schilsky. Wilson disease: Treatment and prognosis. UpToDate, last updated: Oct 14, 2020.
Weiss, K. H. et al. Outcome and development of symptoms after orthotopic liver transplantation for Wilson disease. Clin. Transplant. 27, 914–922 (2013).
Dhawan A, Taylor RM, Cheeseman P. et al. Wilson’s disease in children: 37-year experience and revised King’s score for liver transplantation. Liver Transplant 2005;11:441–448.
Rachel M Taylor, Yuan Chen, Anil Dhawan, EuroWilson Consortium. Triethylene tetramine dihydrochloride (trientine) in children with Wilson disease: experience at King"s College Hospital and review of the literature/(Eur J Pediatr.2009 Sep;168(9):1061-8
http://www.hgmd.cf.ac.uk/ac/gene.php?gene=ATP7B
Dusek P, Litwin T, Członkowska A. Neurologic impairment in Wilson disease. Ann Transl Med. 2019;7(Suppl 2):S64. doi:10.21037/atm.2019.02.43
Czlonkowska A, Litwin T, Chabik G. Wilson disease: neurologic features. Handb Clin Neurol 2017;142:101-19. 10.1016/B978-0-444-63625-6.00010-0
Kwo PY, Cohen SM, Lim JK. ACG Clinical Guideline: Evaluation of Abnormal Liver Chemistries. Am J Gastroenterol. 2017 Jan;112(1):18-35
Wilson disease: Clinical manifestations, diagnosis, and natural history (https://www.uptodate.com/contents/wilson-disease-clinical-manifestations-diagnosis-and-natural-history?search=wilson%20disease&source=search_result&selectedTitle=1%7E150&usage_type=default&display_rank=1#H65234579), last updated: Jul 31, 2023
Wang H, Zhou Z, Hu J, Han Y, Wang X, Cheng N, Wu Y, Yang R. Renal impairment in different phenotypes of Wilson disease. Neurol Sci. 2015 Nov;36(11):2111-5
Güngör Ş, Selimoğlu MA, Varol Fİ. Nutritional assessment of children with Wilson"s disease: single center experience. Turk Pediatri Ars. 2019 Dec 25;54(4):246-255
Gaffney D, Fell GS, O"Reilly DS. ACP Best Practice No 163. Wilson"s disease: acute and presymptomatic laboratory diagnosis and monitoring. J Clin Pathol. 2000 Nov;53(11):807-12. doi: 10.1136/jcp.53.11.807
Вялова Н.В. «Клинические и молекулярно-генетические особенности гепатолентикулярной дегенерации, оптимизация диагностики и динамического наблюдения», диссертация, 2021г
Бикбавова Г.Р., Хомутова Е.Ю, Третьякова Т.В., и др «Диагностические особенности семейного случая Болезни Вильсона-Коновалова» — «Трудный пациент». — 2017 — № 4-5 - т.15.- с.32-35
EASL Clinical Practice Guidelines: Autoimmune hepatitis. J Hepatol 2015; 63:971–1004. 20
Costa da M., Spitz M., Bacheszhi L., et al. Cоrrelation to pretreatment and posttreatment brain MRI//Neuroradiology.-2009.-Vol.21.-P. 627-633
Gilroy Richard K. Wilson Disease. Updated: Feb 14, 2019 (https://emedicine.medscape.com/article/183456-overview)
Tinaz S, Arora J, Nalamada K, Vives-Rodriguez A, Sezgin M, Robakis D, Patel A, Constable RT, Schilsky ML. Structural and functional brain changes in hepatic and neurological Wilson disease. Brain Imaging Behav. 2020 Nov 26
Prashanth LK, Sinha S, Taly AB, A Mahadevan, Vasudev MK, Shankar SK. Spectrum of epilepsy in Wilson"s disease with electroencephalographic, MR imaging and pathological correlates. J Neurol Sci. 2010 Apr 15;291(1-2):44-51. doi: 10.1016/j.jns.2010.01.007
Litwin T, Dusek P, Czlonkowska A. Neurological manifestations in Wilson’s disease –possible treatment options for symptoms. Expert Opinion on Orphan Drugs 2016;4:719-28
Litwin T, Dusek P, Czlonkowska A. Symptomatic treatment of neurologic symptoms in Wilson disease. Handb Clin Neurol 2017;142:211-23
Holscher S, Leinweber B, Hefter H, et al. Evaluation of the symptomatic treatment of residual neurological symptoms in Wilson disease. Eur Neurol. 2010;64(2):83–87
Hedera P. Treatment of Wilson’s disease motor complications with deep brain stimulation. Ann N Y Acad Sci. 2014;1315:16–23
Brewer G.J.Fink J.K.Hedera P.Diagnosis and treatment of Wilson"s disease.Semin Neurol. 1999; 19: 261-270
Reuner U, Dinger J. Pregnancy and Wilson disease: management and outcome of mother and newborns-experiences of a perinatal centre. Ann Transl Med. 2019 Apr;7(Suppl 2):S56
Teive H.A., Kluppel L.E., Munhoz R.P. Jaw-opening oromandibular dystonia secondary to Wilson"s disease treated with botulinum toxin type A. Arq Neuropsiquiatr. 2012;70:407–409
Lee S.Y., Yang H.E., Yang H.S. Neuromuscular electrical stimulation therapy for dysphagia caused by Wilson"s disease. Ann Rehabil Med. 2012;36:409–413
Rodriguez-Castro K, Hevia-Urrutia F, Sturniolo G. Wilson"s disease: A review of what we have learned. World Journal Of Hepatology [serial on the Internet]. (2015, Dec 18), [cited April 8, 2016]; 7(29): 2859-2870.
Wilson Disease Association (WDA) International [Internet]. Wilson Disease Association (WDA) International. [cited 2016Mar30]. Retrieved from: http://www.wilsonsdisease.org/
Brewer G, Askari F. Wilson"s disease: clinical management and therapy. Journal Of Hepatology [serial on the Internet]. (2005), [cited April 8, 2016]; 42 Suppl(1): S13-S21.
van den Berg-Emons R, van Ginneken B, Nooijen C, Metselaar H, Tilanus H, Kazemier G et al. Fatigue After Liver Transplantation: Effects of a Rehabilitation Program Including Exercise Training and Physical Activity Counseling. Physical Therapy. 2014;94(6):857-865
Медицинские противопоказания к проведению профилактических прививок препаратами национального календаря прививок: Методические указания.—М.: Федеральный центр госсанэпиднадзора Минздрава России, 2002.— 16 с.
«Методические рекомендации по выявлению, расследованию и профилактике побочных проявлений после иммунизации» (утв. Минздравом России 12.04.2019)
Kathawala M, Hirschfield GM. Insights into the management of Wilson"s disease. Therap Adv Gastroenterol. 2017 Nov;10(11):889-905, UpToDate.com
Przybyłkowski A, Szeligowska J, Januszewicz M, Raszeja-Wyszomirska J, Szczepankiewicz B, Nehring P, Górnicka B, Litwin T, Członkowska A. Evaluation of liver fibrosis in patients with Wilson"s disease. Eur J Gastroenterol Hepatol. 2021 Apr 1;33(4):535-540
Stefanescu AC, Pop TL, Stefanescu H, Miu N. Transient elastography of the liver in children with Wilson"s disease: Preliminary results. J Clin Ultrasound. 2016 Feb;44(2):65-71
Hoppe B, Neuhaus T, Superti-Furga A, Forster I, Leumann E. Hypercalciuria and nephrocalcinosis, a feature of Wilson"s disease. Nephron. 1993;65(3):460-2
Di Stefano V, Lionetti E, Rotolo N, La Rosa M, Leonardi S. Hypercalciuria and nephrocalcinosis as early feature of Wilson disease onset: description of a pediatric case and literature review. Hepat Mon. 2012 Aug;12(8): e6233
Schaefer M. et al. Coagulation Parameters in Wilson Disease //Journal of Gastrointestinal & Liver Diseases. – 2015. – Т. 24. – №. 2.
Doering E. J., Savage R. A., Dittmer T. E. Hemolysis, coagulation defects, and fulminant hepatic failure as a presentation of Wilson"s disease //American Journal of Diseases of Children. – 1979. – Т. 133. – №. 4. – С. 440-441
Mak C. M., Lam C. W., Tam S. Diagnostic accuracy of serum ceruloplasmin in Wilson disease: determination of sensitivity and specificity by ROC curve analysis among ATP7B-genotyped subjects //Clinical chemistry. – 2008. – Т. 54. – №. 8. – С. 1356-1362
Yüce A. et al. Evaluation of diagnostic parameters of Wilson"s disease in childhood //Indian journal of gastroenterology: Official journal of the Indian Society of Gastroenterology. – 2003. – Т. 22. – №. 1. – С. 4-6.
Da Costa C. M. et al. Value of urinary copper excretion after penicillamine challenge in the diagnosis of Wilson"s disease //Hepatology. – 1992. – Т. 15. – №. 4. – С. 609-615.
Lopez-Sanroman A. et al. Stimulated urinary copper excretion in the diagnosis of Wilson"s disease //Gastroenterologia y hepatologia. – 2015. – Т. 38. – №. 7. – С. 467-467
Gromadzka G. et al. p. H1069Q mutation in ATP7B and biochemical parameters of copper metabolism and clinical manifestation of Wilson"s disease //Movement disorders. – 2006. – Т. 21. – №. 2. – С. 245-248.
Németh D. et al. Clinical use of next-generation sequencing in the diagnosis of Wilson’s disease //Gastroenterology research and practice. – 2016. – Т. 2016.
Okada T. et al. Mutational analysis of ATP7B and genotype–phenotype correlation in Japanese with Wilson"s disease //Human mutation. – 2000. – Т. 15. – №. 5. – С. 454-462.
Roh J. K. et al. Initial and follow‐up brain MRI findings and correlation with the clinical course in Wilson"s disease //Neurology. – 1994. – Т. 44. – №. 6. – С. 1064-1064.
Sinha S. et al. Sequential MRI changes in Wilson"s disease with de-coppering therapy: a study of 50 patients //The British journal of radiology. – 2007. – Т. 80. – №. 957. – С. 744-749.
Dening T. R., Berrios G. E., Walshe J. M. Wilson"s disease and epilepsy //Brain. – 1988. – Т. 111. – №. 5. – С. 1139-1155.
Meenakshi-Sundaram S. et al. Cardiac involvement in Wilson’s disease—an electrocardiographic observation //JAPI. – 2004. – Т. 52.
Buksińska-Lisik M. et al. Cardiac assessment in Wilson’s disease patients based on electrocardiography and echocardiography examination //Archives of medical science: AMS. – 2019. – Т. 15. – №. 4. – С. 857.
Karakurt C. et al. Strain and strain rate echocardiography in children with Wilson"s disease //Cardiovascular journal of Africa. – 2016. – Т. 27. – №. 5. – С. 307-314.
Appenzeller‐Herzog C. et al. Comparative effectiveness of common therapies for Wilson disease: A systematic review and meta‐analysis of controlled studies //Liver International. – 2019. – Т. 39. – №. 11. – С. 2136-2152.
Salivary Glands - New Approaches in Diagnostics and Treatment, 2019. eBook (PDF) ISBN: 978-1-83881-358-1.
Детская гепатология: [монография] / под ред. Б.С. Каганова» — книга автора Багаева М.Э.. Издано: (2009).
Kulkarni A, Sharma VK. Wilson’s disease. In: Quah SR, editor. International encyclopedia of public health, vol. 369. Amsterdam: Elsevier; 2016. p. 424–33. https:// doi.org/10.1016/B978-0-12-803678-5.00495-1. 14 Mulligan & Bronstein
Cheung KS, Seto WK, Fung J, et al. Epidemiology and natural history of Wilson’s disease in the Chinese: A territory-based study in Hong Kong between 2000 and 2016. World J Gastroenterol 2017;23(43):7716–26
Poujois A, Woimant F, Samson S, et al. Characteristics and prevalence of Wilson’s disease: A 2013 observational population-based study in France. Clin Res Hepatol Gastroenterol 2018;42(1):57–63]
Gromadzka G, Wierzbicka D, Litwin T, Przybyłkowski A. Difference in iron metabolism may partly explain sex-related variability in the manifestation of Wilson"s disease. J Trace Elem Med Biol. 2020 Dec;62:126637
Gromadzka G, Wierzbicka D, Litwin T, Przybyłkowski A. Iron metabolism is disturbed and anti-copper treatment improves but does not normalize iron metabolism in Wilson"s disease. Biometals. 2021 Apr;34(2):407-414. doi: 10.1007/s10534-021-00289-x
Dusek P, Hofer T, Alexander J, Roos PM, Aaseth JO. Cerebral Iron Deposition in Neurodegeneration. Biomolecules. 2022 May 17;12(5):714
Schilsky ML, Roberts EA, Bronstein JM, Dhawan A, Hamilton JP, Rivard AM, Washington MK, Weiss KH, Zimbrean PC. A multidisciplinary approach to the diagnosis and management of Wilson disease: 2022 Practice Guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology. 2022 Dec 7. doi: 10.1002/hep.32801
Scheinberg IH, Jaffe ME, Sternlieb I. The use of trientine in preventing the effects of interrupting penicillamine therapy in Wilson"s disease. N Engl J Med. 1987;317:209–13, Saito H, Watanabe K, Sahara M, Mochizuki R, Edo K, Ohyama Y. Triethylene‐tetramine (trien) therapy for Wilson"s disease. Tohoku J Exp Med. 1991;164:29–35
Taylor RM, Chen Y, Dhawan A, on behalf of the EuroWilson Consortium. Triethylene tetramine dihydrochloride (trientine) in children with Wilson disease: experience at King"s College Hospital and review of the literature. Eur J Pediatr. 2009;168:1061–8
Mayr T, Ferenci P, Weiler M, Fichtner A, Mehrabi A, Hoffmann GF, et al. Optimized trientine‐dihydrochloride therapy in pediatric patients with Wilson disease: is weight‐based dosing justified? J Pediatr Gastroenterol Nutr. 2021;72:115–22
Annu Aggarwal and Mohit Bhatt /Advances in Treatment of Wilson Disease/ Tremor Other Hyperkinet Mov (N Y). 2018; 8: 525.
Rachel M Taylor, Yuan Chen, Anil Dhawan, EuroWilson Consortium. Triethylene tetramine dihydrochloride (trientine) in children with Wilson disease: experience at King"s College Hospital and review of the literature/(Eur J Pediatr.2009 Sep;168(9):1061-8
Неврология: справочник практ. врача / О.С.Левин, Д.Р.Штульман. – 12‑е изд. – М. : МЕДпресс-информ, 2019. – 879 с.
Mariño Z, Molera-Busoms C, Badenas C, Quintero-Bernabeu J, Torra M, Forns X, Artuch R. Benefits of using exchangeable copper and the ratio of exchangeable copper in a real-world cohort of patients with Wilson disease. J Inherit Metab Dis. 2023;46(5):982-991. doi: 10.1002/jimd.12639
Członkowska A, Litwin T, Dusek P, Ferenci P, Lutsenko S, Medici V, Rybakowski JK, Weiss KH, Schilsky ML. Wilson disease. Nat Rev Dis Primers. 2018;4(1):21. doi: 10.1038/s41572-018-0018-3
Fernando M, van Mourik I, Wassmer E, Kelly D. Wilson disease in children and adolescents. Arch Dis Child. 2020;105(5):499-505. doi: 10.1136/archdischild-2018-315705
Kaplan JH, Maryon EB. How mammalian cells acquire copper: an essential but potentially toxic metal. Biophys J. 2016 Jan 5;110(1):7-13. doi: 10.1016/j.bpj.2015.11.025
Rochford G, Molphy Z, Kavanagh K, McCann M, Devereux M, Kellett A, et al. Cu(ii) phenanthroline-phenazine complexes dysregulate mitochondrial function and stimulate apoptosis. Metallomics 2020;12:65–78. doi: 10.1039/c9mt00187e
Shao J, Li M, Guo Z, Jin C, Zhang F, Ou C, et al. TPP-related mitochondrial targeting copper (II) complex induces p53-dependent apoptosis in hepatoma cells through ROS-mediated activation of Drp1. Cell Commun Signal 2019;17:149. doi: 10.1186/s12964- 019-0468-6
Chanpong A, Dhawan A.Wilson disease in children and young adults - State of the art. Saudi J Gastroenterol. 2022;28(1):21-31. doi: 10.4103/sjg.sjg_501_21
Kerkar N, Rana A. Wilson Disease in Children. Clin Liver Dis. 2022;26(3):473-488. doi: 10.1016/j.cld.2022.03.008
Członkowska A, Litwin T, Chabik G. Wilson disease: neurologic features. Handb Clin Neurol. 2017;142:101-119. doi: 10.1016/B978-0-444-63625-6.00010-0
Zimbrean PC, Schilsky ML. Psychiatric aspects of Wilson disease: a review. Gen Hosp Psychiatry 2014;36:53–62
Litwin T, Dusek P, Szafrański T, et al. Psychiatric manifestations in Wilson’s disease: possibilities and difficulties for treatment. Ther Adv Psychopharmacol 2018;8:199–211
Pfeiffenberger J, Beinhardt S, Gotthardt DN, Haag N, Freissmuth C, Reuner U, Gauss A, Stremmel W, Schilsky ML, Ferenci P, Weiss KH. Pregnancy in Wilson"s disease: Management and outcome. Hepatology. 2018;67(4):1261-1269. doi: 10.1002/hep.29490
Malik A, Khawaja A, Sheikh L. Wilson"s disease in pregnancy: Case series and review of literature. BMC Res Notes. 2013;6:421. doi: 10.1186/1756-0500-6-421