При НТ1 обычно беременность и перинатальный период протекают нормально, и ребенок при рождении выглядит здоровым. Семейный анамнез может быть отягощен, в семьях могут быть случаи ранней детской смерти от кровоизлияний в мозг или острых кровотечений, расцененных как сепсис, внутриутробные инфекции, синдром диссименированного внутрисосудистого свертывания крови.
Острая форма наследственной НТ1 (НТ1А) дебютирует в первые 2-3 месяца жизни и характеризуется главным образом тяжелой печеночной недостаточностью, гепато- и спленомегалией, нарушенной свертываемостью крови и гипогликемией, приводящими к смерти в первые месяцы жизни. Дисфункция почечных канальцев сопровождается синдромом Фанкони и рахитом [2, 8]. Пациентам довольно часто ставят ошибочные диагнозы - сепсис, цитомегаловирусный гепатит, фетальный гепатит. У детей с НТ1А выражена интоксикация, гипогликемия, лейкоцитоз, возможен асцит, динамическая непроходимость, парез кишечника. При этом отмечается повышенный уровень сукцинилацетона (≥ 10–100 раз), альфа-фетопротеина (АФП) (≥ 100–1000 раз). Цитолиз обычно не превышает трех-четырех норм, умеренная гипербилирубинемия в пределах 100–150 мкмоль/л, в равном соотношении прямой и непрямой билирубин. Причиной летального исхода при естественном течении НТ1А обычно становится острая печеночная недостаточность и катастрофическое кровотечение на ее фоне. Диагноз затрудняется такими сопутствующими состояниями, как внутриутробная инфекция, неонатальный гепатит, сепсис.
При острой форме НТ1 на первый план в клинической картине выступают симптомы интоксикации (высокая температура, повышенная возбудимость, по мере нарастания симптоматики наступает нарушение уровня сознания вплоть до комы), а также симптомы поражения печени (увеличение печени, может быть в сочетании с увеличением селезенки, асцит, желтуха — редко), гастроинтестинальный синдром (срыгивания после каждого кормления), тяжелый геморрагический синдром (мелена, кровавая рвота, гематурия, экхимозы, петехии, носовые кровотечения), специфический запах мочи (запах «вареной капусты»). С 3-х недельного возраста , если имеют место частые срыгивания и рвота, остановка в прибавке массы тела, выраженная мышечная гипотония, сонливость, неспецифические симптомы интоксикации, серость кожи, увеличение размеров живота за счет асцита, гепато-спленомегалии, динамическая непроходимость (парез кишечника) при наличии жидких каловых масс, с кровью или без, возможны отеки , пастозность стоп, в лабораторных данных гипогликемия, гипокоагуляция, лейкоцитоз, цитолиз обычно не выше 3 норм, повышен уровень ГГТП, при этом билирубиновый обмен обычно не нарушен, т.е. имеет место холестаз без желтухи, АФП повышен значительно ( при норме до 60 нг/мл у детей до 5 мес.) достигал 1 млн. нг/мл, ребенок подлежит селективному скринингу на болезни обмена , в том числе на НТ1.
При хронической форме (НТ1В) отмечается другая клиническая картина, болезнь развивается после годовалого возраста, чаще в 3-4 года. НТ1В изначально менее агрессивна, но заболевание неуклонно прогрессирует и может сопровождаться приступами острой метаболической декомпенсации. При этой форме наблюдаются различной степени выраженности поражения печени, почечные канальциевые дисфункции, рахитоподобные изменения скелета, обусловленные гипофосфатемией и гиперфосфатурией, кардиомиопатия, неврологические кризы, сходные с порфирическими (болезненные парестезии, артериальная гипертония, тахикардия, паралитическая непроходимость кишечника, иногда прогрессирующие параличи), гипогликемия, связанная с гиперплазией β-клеток поджелудочной железы и гиперсекрецией инсулина. Неврологические кризы обычно длятся 1–7 дней, после чего наступает период восстановления.
У пациентов наблюдается нарушение функции реабсорбции в почечных канальцах, приводящее к синдрому Фанкони, ацидозу, генерализованной аминоацидурии, резистентному к витамину D гипофосфатемическому рахиту и задержке роста. В ряде случаев у ребенка наблюдается рахит с деформациями конечностей, столь выраженной, что дети перестают ходить. В этом же возрасте диагностируют цирроз, гепатоспленомегалию, могут быть носовые кровотечения, гипопротеинемические отеки. НТ1В характеризуется постепенными изменениями печени, приводящими к циррозу и развитию гепатоцеллюлярной карциномы. Фактически риск развития гепатоцеллюлярной карциномы при этом заболевании считается самым высоким среди всех нарушений обмена веществ [17, 18]. Ранее сообщалось, что гепатоцеллюлярная карцинома формируется у 37% пациентов с наследственной НТ1 старше 2 лет [19], но последующие исследования в Скандинавии [20] и Квебеке [17] показали более низкую частоту гепатоцеллюлярной карциномы (~15%), вероятно, вследствие трансплантации печени и улучшения способов лечения. Гепатоцеллюлярная карцинома у пациентов с наследственной тирозинемией 1-го типа развивается в более раннем возрасте, чем при других наследственных болезнях обмена (часто до 5 лет) [18]. Цитолиз минимальный (в пределах трех норм). Уровень АФП повышен в пределах 1000 нг/дл, сукцинилацетона – не более чем в 20 раз [9].
Подострая и хроническая формы болезни встречаются примерно у 20–25% больных с дебютом в 4-12 мес, клиническая манифестация которой провоцируется внешними факторами. Такими тригерными факторами являются лихорадочные состояния, инфекции, белковая нагрузка, голодание и другие гиперкатаболические состояния. У пациентов обнаруживается прогрессирующее поражение печени с гепато- или гепатоспленомегалией (цирроз, печеночная недостаточность), задержка роста, признаки рахита, мышечная гипотония, коагулопатия. Характерно отставание показателей физического развития. Неврологические симптомы нередко включают гидроцефальную конфигурацию черепа, симптом Грефе, задержку двигательного и психического развития. Могут наблюдаться неврологические кризы (слабость в конечностях, параличи конечностей и диафрагмы, рвота, аутоагрессия и др.), которые в ряде случаев служат поводом для ошибочного диагноза — myasthenia gravis. Неврологические изменения непостоянны. Нередко описывают гиперпигментацию кожи.
Следует отметить, что эта классификация довольно условна, поскольку некоторые дети с острой декомпенсацией в течение первого года жизни выживают и их форма становится «хронической», а пациенты с «хронической» формой остаются в группе риска по развитию метаболических кризов. При этом, возраст дебюта является важным прогностическим показателем. До появления патогенетического лечения в течение 1-го года умерли 60 % детей с дебютом заболевания в возрасте до 2 месяцев, 23 % при дебюте в возрасте от 2 до 6 месяцев и только 4 % детей дебютировавших после 6 месяцев [21]. В некоторых регионах РФ проводятся программы по массовому обследованию на HT1. Следует отметить, что до 4 дней жизни у доношенных и до 7 дней жизни у недоношенных новорожденных, неонатальный скрининг будет неэффективным, так как в крови у пациентов с HT1 еще не накапливаются высокие концентрации сукцинилацетона [31, 36].
Печеночные проявления тирозинемии I типа
Основные клинические проявления HT1 связаны с поражением печени. Могут наблюдаться острая печеночная недостаточность, цирроз печени и гепатоцеллюлярная карцинома [21]. По данным международных исследований 69% смертей были связаны с печеночной недостаточностью и кровотечением, а 16% - с гепатоцеллюлярной карциномой. Синтетическая функция нарушена у большинства пациентов с HT1. Уровень факторов свертывания заметно снижен при HT1, и у некоторых пациентов именно нарушения коагуляции являются ведущими, другие признаки поражения печени выражены незначительно, поэтому пациентов обследуют на предмет первичной гематологической проблемы [22]. Протромбиновое и частичное тромбопластиновое время может быть чрезвычайно продолжительным даже у клинически здоровых детей с НТ1, выявленных при скрининге. Применение витамина К только на короткое время может улучшить профиль коагуляции. Уровни трансаминаз в сыворотке варьируют и могут быть нормальными или незначительно повышенными, хотя в некоторых кризисных ситуациях могут наблюдаться уровни более 1000 МЕ / л, что указывает на более существенную степень повреждения гепатоцитов, и как правило заставляет предпринять шаги к экстренной трансплантации печени. Желтуха редко встречается на ранних стадиях гепаторенальной тирозинемии. Тогда как, гамма-глютамилтрансфераза обычно повышена более чем в 2 раза, и соответствует диссоциированному холестазу.
Печеночные кризы могут быть спровоцированы инфекциями и другими катаболическими стрессами как у младенцев, так и у детей старшего возраста. Ребенок раздражителен, ослаблен и часто лихорадит. Часто встречаются асцит, желтуха и желудочно-кишечные кровотечения. Может ощущаться запах «вареной капусты», но запах наблюдается не часто - примерно в 15% случаев. Имеется гепатомегалия различной степени. Криз сопровождается выраженной потливостью, гипергидроз зачастую объясняется течением острого рахита, что не исключает, а лишь подтверждает HT1. Тремор, гипергидроз и резкая слабость как проявление гипогликемии в период криза встречается чаще, чем вне его. Во время кризов наблюдаться повышение уровня тирозина, метионина, пролина и часто других аминокислот в плазме. Печеночные трансаминазы могут резко повышаться, и может развиваться гипербилирубинемия, что соответствует распаду гепатоцитов. В этот период может усилиться дисфункция почечных канальцев. Хотя многие кризы разрешаются спонтанно, при уменьшении белковой нагрузки, адекватного парентерального питания и детоксикационной терапии, и некоторые прогрессируют до полной печеночной недостаточности и печеночной энцефалопатии.
Между кризами у большинства пациентов наблюдается гепатомегалия легкой или средней степени тяжести. У некоторых пациентов с формирующимся циррозом или гепатомегалией с нарушением гемодинамики и повышением резистентности паренхимы (по ДГ сосудов печени) отмечается спленомегалия. Печеночные трансаминазы в норме или немного повышены, а уровень билирубина в норме. Альфа-фетопротеин повышен (10–1 000 000 нг/мл; в норме <10). Клинически уровни α-фетопротеина в сыворотке ориентировочно отражают агрессивность заболевания, как и уровни метионина в плазме. Гипербилирубинемия и гипераммониемия являются поздними признаками печеночной недостаточности
Цирроз, вероятно, в конечном итоге развивается у всех пациентов с HT1, и риск гепатоцеллюлярной карциномы чрезвычайно высок [19]. С каждым годом без лечения болезни риск развития ГЦК увеличивается с 14 до 75%. Раннее, до месячного возраста, начало патогенетичсекой терапии снижает риск ГЦК до 1%, и необходимость в трансплантации печени уменьшается с 71% до 26% [15, 23]. Важно, что далеко не каждый случай ГЦК возникает на фоне цирроза. Так в обзоре R.Khanna, 2018, включающем 15 исследований, средняя частота ГЦК у детей составила 0,41 на 1000000, у взрослых -0,8 на миллион населения [24]. Частота ГЦК при НТ1 превышает в 40 раз ее частоту при других метаболических и инфекционных заболеваниях печени. Риск развития ГЦК при НТ1 возникает при отсутствии терапии нитизиноном и началом терапии позже 1 месячного возраста [25]. Тем не менее, даже при длительном периоде без лечения, часть детей благодарно отвечают на терапию – восстанавливаются структура печени, купируется тубулопатия, восстанавливается минеральная плотность костей и электролитный баланс. Отмечены случаи, когда пациенты были направлены на трансплантацию с предварительной подготовкой NTBC (2-(2-нитро-4-трифлюорометилбензоил)-1,3-циклогексанедион (нитизинон)) для улучшения обменных процессов, курсом на 3 мес., а по прошествии этого периода, показания к трансплантации исчезали. У некоторых детей продолжалась консервативная терапия, стабилизация и/или регресс цирроза.
Большинство инструментальных методов исследования (КТ, УЗИ, ангиография) не позволяют надежно дифференцировать доброкачественные и злокачественные узелковые образования. Для КТ-обнаружения важно выполнять томографию с контрастом и без него, потому что контрастирование может скрывать одни узелки, но выявить другие. Чувствительность УЗИ по крайней мере такая же, как у КТ, но оно во многом зависит от опыта врача. Следует отметить, что поскольку премедикация барбитуратами может спровоцировать порфирийно-подобные кризы, лучше избегать ее использования у пациентов, не получавших NTBC [26].
Неврологические кризисы тирозинемии I типа
Неврологические кризы гепаторенальной тирозинемии — это острые эпизоды периферической невропатии. Клинически кризы делятся на две фазы: (1) активный период с преобладанием болезненных парестезий, вегетативных признаков (например, гипертония, тахикардия, кишечная непроходимость) и иногда прогрессирующего паралича и (2) период восстановления после паралитического криза. Из 48 пациентов франко-канадского происхождения у 20 (42 процента) были описаны неврологические кризы [27]. Периферическая гиперпарестезия проявляется с раннего возраста -младенец отказывается находится на руках у матери, беспокоится при прикосновениях, поглаживаниях, предпочитает находится в кроватке. Болезненные кризы наиболее частый неврологический вариант [28]. Во время продрома, который часто возникает после незначительной инфекции с анорексией и рвотой, ребенок раздражителен и менее активен, чем обычно. Затем у ребенка появляется сильная боль, часто в ногах. Часто пациенты принимают позицию крайнего гиперэкстензии туловища и шеи, которые могут быть ошибочно приняты за опистотонус или менингизм. Гипертонус можно принять за тонические судороги, но на самом деле пациенты находятся в сознании. Также могут наблюдаться настоящие судороги, часто в сочетании с тяжелой гипонатриемией [27]. Кризы могут быть настолько мучительными, что сопровождаются самоповреждениями [27]. Важно отметить, что умственное развитие детей с НТ1 является нормальным и что во время кризисов уровень их сознания не снижается.
Активная фаза обычно длится от 1 до 7 дней. В очень редких случаях во время кризов искусственная вентиляция легких была необходима из-за дыхательной слабости, в одном случае на срок более 3 месяцев [27]. Часто возникают рвота и кишечная непроходимость, которые могут усложнить режим питания. Артериальная гипертония и стойкая тахикардия характерны для ранней стадии неврологического криза. Могут также возникнуть выраженная гипонатриемия, гипофосфатемия и гипокалиемия [27].
Следует отметить, что у 30 российских пациентов не наблюдали подобной острой и тяжелой симптоматики. Неврологические кризы являются основной причиной заболеваемости у пациентов, не получавших NTBC. По данным международного опроса, 10% смертей при НТ1 произошли во время неврологических кризисов. Детей с НТ1 и признаками, указывающими на приближающийся неврологический кризис, следует госпитализировать для постоянного наблюдения за респираторной функцией во время острой фазы.
Патология почек при тирозинемии I типа
У пациентов с НТ1 почти всегда присутствует поражение почек различной степени, от легкой дисфункции канальцев до явной почечной недостаточности. Поражение почек соответствуют вторичному синдрому Фанкони, полному или неполному : снижение уровня кальция в крови; снижение уровня фосфора в крови; повышение уровня щелочной фосфатазы; развитие метаболического ацидоза, нарушение реабсорбции бикарбонатов в проксимальных канальцах; может быть нормальная экскреция кальция с мочой при гипокальцемии; повышение клиренса фосфатов мочи, всасывание фосфатов в кишечнике не страдает; глюкозурия (20-30 г/л и выше); развитие генерализованной гипераминоацидурии; нарушение функций аммониоацидогенеза — снижение титрационной кислотности, повышение рН мочи больше 6,0; развитие гипокалиемии.
Тяжесть дисфункции проксимальных канальцев различна и может резко усугубляться в периоды декомпенсации. Гипофосфатемический рахит является основным клиническим признаком дисфункции почечных канальцев при НТ1. У некоторых детей с НТ1 наблюдается почечный канальцевый ацидоз. Генерализованная аминоацидурия встречается чаще, чем глюкозурия. Потеря фосфата с мочой, вероятно, является основным механизмом рахита у этих пациентов, о чем свидетельствуют частые проявления гипофосфатемии и отсутствие гипокальциемии или гипоплазии эмали зубов, что может свидетельствовать о патогенезе, связанном с витамином D [29].
Скелетные изменения при тирозинемии I типа
До появления возможностей патогенетического лечения пациенты с НТ1 имели грубые многоплоскостные деформации скелета, патологические переломы, утрированные проявления рахита в виде гиперплазии хрящей (четки и браслеты), башенного черепа, гипергидроза, синдрома Хвостека, раздражительности, потливости, ломкости ногтей, зубов. При проведении рентгенографии выявляют снижение костного возраста и остеопению, вплоть до остеопороза, расширение дистальных эпифизов трубчатых костей, деформация и истончение кортикального слоя пястных и плюсневых костей, предплечий, голеней, бедер, саблевидные деформации всех костей на которые была нагрузка – если ходил – нижних конечностей, если ползал - и бедер, и плеч, и предплечий. Наблюдаются деформация грудной клетки, ключиц, выраженная гаррисонова борозда, дисплазия и подвывих тазобедренных суставов.
Диагноз наследственной НТ1 следует предполагать при наличии вальгусных (чаще) или варусных искривлений конечностей с сопутствующей гепатомегалией. Некоторые больные до 2-3 лет самостоятельно передвигаются, хотя резистентность к нагрузкам у них низкая, а затем перестают ходить, что не редко является поводом для обращения к врачам. Это довольно типично для пациентов из многодетных, социально неблагополучных или плохо говорящих по-русски семей из сельской местности, горных аулов, удаленных районов, где нет специалистов.
Другие клинические проявления и дифференциальная диагностика тирозинемии тип 1
Клинически значимая гипертрофическая кардиомиопатия была зарегистрирована у трех детей с тирозинемией, и у одного она стала причиной смерти [30].
Гипогликемия - симптом типичный для младенцев до года с тяжелым течением заболевания, сопровождавшемся печеночной недостаточностью. Поэтому следует проводить дифференциальный диагноз с гликогенозами, нарушениями бета окисления жирных кислот.
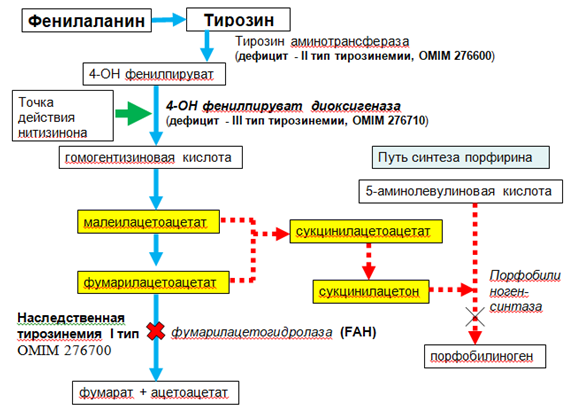
Повышение уровня тирозина также может наблюдаться при других формах наследственной тирозинемии, которые отличаются по спектру клинических проявлений. Недостаточность тирозинаминотрансферазы – первого фермента пути катаболизма тирозина – приводит к развитию наследственной тирозинемии 2-го типа (OMIM 276600), также называемой синдромом Ричнера–Ханхарта. Тирозинемия 2-го типа сопровождается повышением уровня тирозина в крови и моче. Клинический фенотип включает умственную отсталость, болезненные высыпания на роговице, фотофобию, кератит и болезненный пальмоплантарный гиперкератоз [4]. Мутации, приводящие к снижению активности p-гидроксифенилпируватдиоксигеназы – второго фермента катаболизма тирозина, могут способствовать развитию хокинсинурии (OMIM 140350) или наследственной тирозинемии 3-го типа (OMIM 276710).