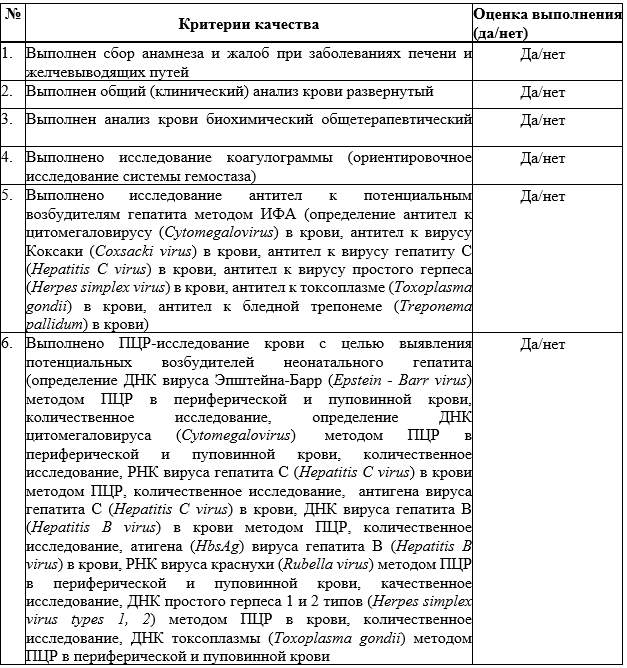
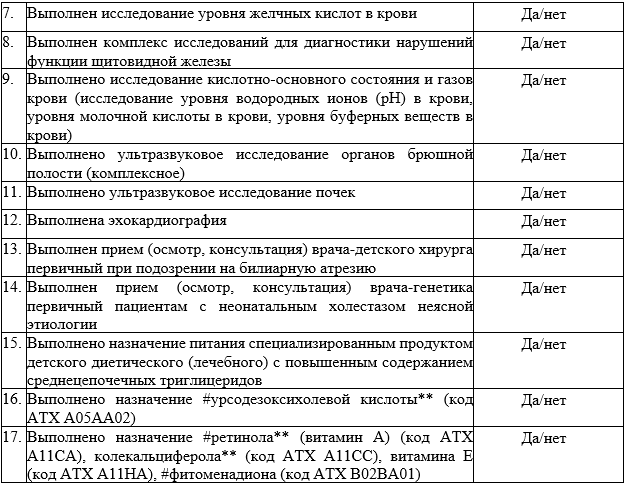
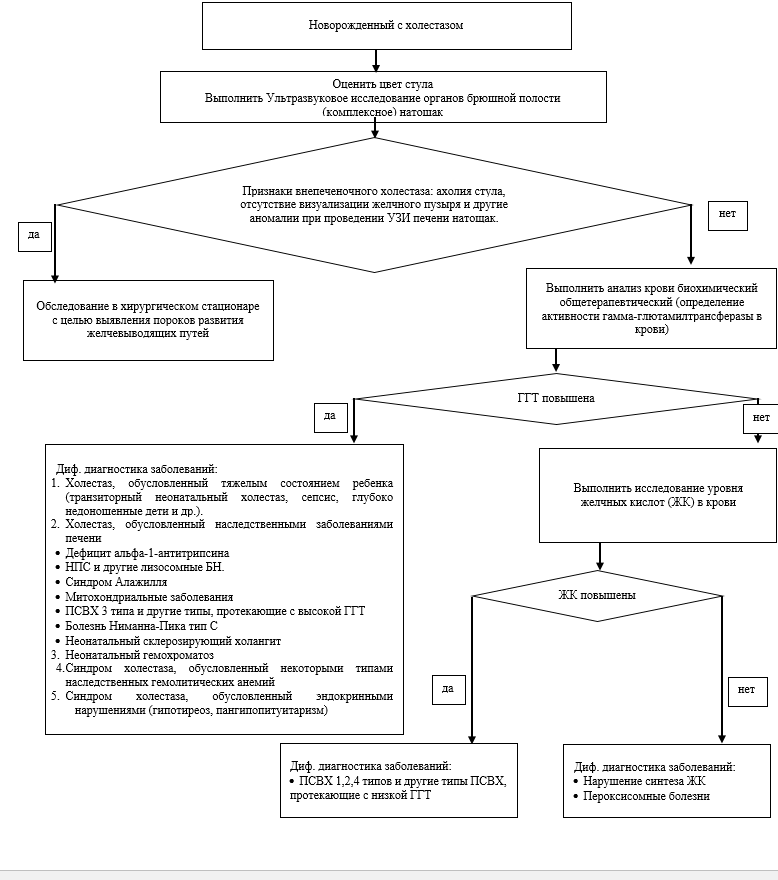
Suchy F.J. Neonatal Cholestasis // Pediatr Rev. 2004. Т. 25, № 11. С. 388–396.
Дегтярева А.В., Мухина Ю.Г., Володин Н.Н. Дифференциальная диагностика и лечение синдрома холестаза у новорожденных детей // Вопросы практической педиатрии. 2007. Т. 2, № 1. С. 55–63.
Lorenzo D’Antiga. Pediatric Hepatology and Liver Transplantation / под ред. D’Antiga L. Cham: Springer International Publishing, 2019. 829 с.
Feldman A.G., Sokol R.J. Neonatal cholestasis // Neoreviews. American Academy of Pediatrics, 2013. Т. 14, № 2. С. 21.
Ghoda M.K. Clinical Casebook Neonatal and Pediatric Liver and Metabolic Diseases. 2021. 278 с.
D’antiga L. Pediatric hepatology and liver transplantation // Pediatric Hepatology and Liver Transplantation. Springer International Publishing, 2019. 1–829 с.
Клинические рекомендации «Нарушения обмена галактозы (Галактоземия)» (возрастная категория взрослые, дети), 2024.
Клинические рекомендации «Наследственная тирозинемия I типа» (возрастная категория взрослые, дети), 2024.
Клинические рекомендации «Болезнь Ниманна-Пика тип С» (возрастная категория взрослые, дети), 2024.
Клинические рекомендации «Кистозный фиброз (муковисцидоз)» (возрастная категория взрослые, дети), 2021.
Клинические рекомендации «Врожденный гипотиреоз у детей », 2024.
Venigalla S., Gourley G.R. Neonatal cholestasis // Seminars in Perinatology. 2004. Т. 28, № 5. С. 348–355.
Abbey P., Kandasamy D., Naranje P. Neonatal Jaundice // The Indian Journal of Pediatrics. 2019. Т. 86, № 9. С. 830–841.
Fischler B., Papadogiannakis N., Nemeth A. Aetiological factors in neonatal cholestasis // Acta Paediatr. 2007. Т. 90, № 1.
Nio M. Introduction to Biliary Atresia // Introduction to Biliary Atresia. Springer Nature, 2021. 1–350 с.
Дегтярева А.В. и др. Билиарная атрезия. Диагностика и лечение. Москва: Прима Принт, 2019. Т. 76.
Клинические рекомендации «Врожденная цитомегаловирусная инфекция», 2023. 62 с.
Spinner N.B., Leonard L.D., Krantz I.D.. Alagille syndrome / под ред. Pagon RA A.M.A.H.W.S.A.A.B.L.B.T.L.N.M.H.S.R.S.K. Seattle: University of Washington, 1993.
Pan Q. и др. A homozygous R148W mutation in Semaphorin 7A causes progressive familial intrahepatic cholestasis // EMBO Mol Med. 2021. Т. 13, № 11.
Luan W. и др. Biallelic loss-of-function ZFYVE19 mutations are associated with congenital hepatic fibrosis, sclerosing cholangiopathy and high-GGT cholestasis // J Med Genet. BMJ Publishing Group, 2021. Т. 58, № 8. С. 514–525.
Amendola M. и др. Pediatric Genetic Cholestatic Liver Disease Overview. 2022. 1993–2024 с.
Zhang J. и др. Low‐GGT intrahepatic cholestasis associated with biallelic USP53 variants: Clinical, histological and ultrastructural characterization // Liver International. 2020. Т. 40, № 5. С. 1142–1150.
Li Q., Sun Y., Van Ijzendoorn S.C.D. biology A Link between Intrahepatic Cholestasis and Genetic Variations in Intracellular Trafficking Regulators. 2021.
Amirneni S. и др. Molecular overview of progressive familial intrahepatic cholestasis // World J Gastroenterol. 2020. Т. 26, № 47. С. 7470–7484.
Goldberg A., Mack C.L. Inherited Cholestatic Diseases in the Era of Personalized Medicine // Clinical Liver Disease. John Wiley and Sons Inc, 2020. Т. 15, № 3. С. 105–109.
Baker A. и др. Systematic review of progressive familial intrahepatic cholestasis // Clin Res Hepatol Gastroenterol. 2019. Т. 43, № 1. С. 20–36.
Davit-Spraul A. и др. Progressive familial intrahepatic cholestasis // Orphanet J Rare Dis. 2009. Т. 4, № 1. С. 25–36.
Bilal H. и др. Neonatal Cholestasis: The Changing Etiological Spectrum in Pakistani Children // Cureus. 2022.
Shneider B.L. и др. Initial assessment of the infant with neonatal cholestasis—Is this biliary atresia? // PLoS One. 2017. Т. 12, № 5. С. e0176275.
Kowalski A. и др. Choledochal Cyst Excision in Infants—A Retrospective Study // Children. Multidisciplinary Digital Publishing Institute (MDPI), 2023. Т. 10, № 2.
Turnpenny P.D., Ellard S. Alagille syndrome: pathogenesis, diagnosis and management // European Journal of Human Genetics. 2012. Т. 20, № 3.
Garcia M.A. и др. Alagille Syndrome: Cutaneous Manifestations in 38 Children // Pediatr Dermatol. 2005. Т. 22, № 1.
Kamath B.M., Spinner N.B., Rosenblum N.D. Renal involvement and the role of Notch signalling in Alagille syndrome // Nat Rev Nephrol. 2013. Т. 9, № 7. С. 409–418.
Semenova N. и др. Clinical Characterization of Alagille Syndrome in Patients with Cholestatic Liver Disease // Int J Mol Sci. Multidisciplinary Digital Publishing Institute (MDPI), 2023. Т. 24, № 14.
Ipatova M.G. и др. A complex case of progressive familial intrahepatic cholestasis type 2 diagnostics // Pediatria. Journal named after G.N. Speransky. 2017. Т. 96, № 6.
Fawaz R. и др. Guideline for the Evaluation of Cholestatic Jaundice in Infants // J Pediatr Gastroenterol Nutr. 2017. Т. 64, № 1. С. 154–168.
Ranucci G. и др. Diagnostic approach to neonatal and infantile cholestasis: A position paper by the SIGENP liver disease working group // Digestive and Liver Disease. 2022. Т. 54, № 1. С. 40–53.
Мухина Ю.Г., Дегтярева А.В. Синдром холестаза у детей первых месяцев жизни. Часть 1 // Доктор. Ру. 2010. С. 22–27.
De Bruyne R. et al. Clinical practice: neonatal cholestasis //European journal of pediatrics. – 2011. – Т. 170. – С. 279-284..
Raquel B Pinto et al. Cholestatic Jaundice with Hypoglycemia as a Manifestation of Congenital Endocrine Disorders: A Case Series // Arch Pediatr. 2022. Т. 7, № 1.
Petrescu A.D. и др. Hypothalamus-Pituitary-Adrenal Dysfunction in Cholestatic Liver Disease // Front Endocrinol (Lausanne). 2018. Т. 9. С. 660.
Chan U. и др. Cholestasis caused by panhypopituitarism and acquired cytomegalovirus infection in a 2-month-old male infant // Medicine. 2017. Т. 96, № 17. С. e6757.
El Qadiry R. и др. Neonatal Cholestasis: A Rare and Unusual Presentation of Pituitary Stalk Interruption Syndrome // Case Rep Endocrinol. 2021. Т. 2021. С. 1–3.
А.В. Болмасова и др. Трудности диагностики врожденного гипопитуитаризма в неонатальном периоде // Неонатология: новости, мнения, обучение. 2017. Т. 2, № 16. С. 81–90.
Souder J.P. и др. Congenital Cytomegalovirus and Hepatic Failure // Pediatric Infectious Disease Journal. 2022. Т. 41, № 2. С. e49–e53.
Teng J. и др. High Rate of Cytomegalovirus Detection in Cholestatic Preterm Infants // Front Pediatr. 2021. Т. 9.
Zhao D. и др. Effects of cytomegalovirus infection on the differential diagnosis between biliary atresia and intrahepatic cholestasis in a Chinese large cohort study // Ann Hepatol. 2021. Т. 23. С. 100286.
Boyer S.G., Boyer K.M. Update on TORCH infections in the newborn infant // Newborn and Infant Nursing Reviews. 2004. Т. 4, № 1. С. 70–80.
Tian C., Ali S.A., Weitkamp J.-H. Congenital Infections, Part I: Cytomegalovirus, Toxoplasma , Rubella, and Herpes Simplex // Neoreviews. 2010. Т. 11, № 8. С. e436–e446.
Shigetomi H. и др. Early Detection and Diagnosis of Neonatal Intrahepatic Cholestasis Caused by Citrin Deficiency Missed by Newborn Screening Using Tandem Mass Spectrometry // Int J Neonatal Screen. 2018. Т. 4, № 1. С. 5.
Mushtaq I. и др. Screening of newborn infants for cholestatic hepatobiliary disease with tandem mass spectrometry Commentary: What is tandem mass spectrometry? // BMJ. 1999. Т. 319, № 7208. С. 471–477.
Zhang H. и др. Expanded newborn screening for inherited metabolic disorders by tandem mass spectrometry in a northern Chinese population // Front Genet. 2022. Т. 13.
Kido J. и др. Improved sensitivity and specificity for citrin deficiency using selected amino acids and acylcarnitines in the newborn screening // J Inherit Metab Dis. 2023.
Nittono H. и др. Navigating cholestasis: identifying inborn errors of bile acid metabolism for precision diagnosis // Front Pediatr. Frontiers Media SA, 2024. Т. 12.
Xiao G. и др. 206,977 newborn screening results reveal the ethnic differences in the spectrum of inborn errors of metabolism in Huaihua, China // Front Genet. Frontiers Media SA, 2024. Т. 15.
Wang D. и др. Disease spectrum, prevalence, genetic characteristics of inborn errors of metabolism in 21,840 hospitalized infants in Chongqing, China, 2017-2022 // Front Genet. Frontiers Media SA, 2024. Т. 15.
Niemi A.-K. Neonatal Presentations of Metabolic Disorders Practice Gaps // Neoreviews. 2021. С. 649–662.
Quelhas P. и др. Protocols of Investigation of Neonatal Cholestasis—A Critical Appraisal // Healthcare (Switzerland). MDPI, 2022. Т. 10, № 10.
Xie S. и др. Genetic alterations and molecular mechanisms underlying hereditary intrahepatic cholestasis // Frontiers in Pharmacology. Frontiers Media SA, 2023. Т. 14.
Hahn J.W. и др. Diagnostic algorithm for neonatal intrahepatic cholestasis integrating single‐gene testing and next‐generation sequencing in East Asia // J Gastroenterol Hepatol. 2024. Т. 39, № 5. С. 964–974.
Verkade H.J. и др. EASL Clinical Practice Guidelines on genetic cholestatic liver diseases // J Hepatol. Elsevier BV, 2024.
Kirchberg I. и др. Distinct neonatal hyperammonemia and liver synthesis dysfunction: case report of a severe MEGDHEL syndrome // Front Pediatr. Frontiers Media SA, 2024. Т. 12.
Колчина А.Н. и др. Диагностика наследственных болезней обмена веществ у новорожденных с синдромом гипераммониемии в дебюте заболевания (пилотное исследование) // Современные технологии в медицине. Privolzhsky Research Medical University, 2021. Т. 13, № 1. С. 59–65.
Соколова Е.В. и др. Клиническое наблюдение ребенка c гипераммониемией, ассоциированной с приемом препарата вальпроевой кислоты // Неонатология: Новости. Мнения. Обучение. Geotar Media Publishing Group, 2023. Т. 11, № 4. С. 36–43.
Дегтярева А.В. и др. Гипераммониемия в практике неонатолога. // Российский вестник перинатологии и педиатрии. National Academy of Pediatric Science and Innovation, 2020. Т. 65, № 6. С. 98–107.
Savy N. и др. Acute pediatric hyperammonemia: current diagnosis and management strategies // Hepat Med. Dove Medical Press Ltd., 2018. Т. Volume 10. С. 105–115.
Napolitano M. и др. Practical approach to imaging diagnosis of biliary atresia, Part 1: prenatal ultrasound and magnetic resonance imaging, and postnatal ultrasound // Pediatr Radiol. 2021. Т. 51, № 2. С. 314–331.
Berauer J. и др. Identification of Polycystic Kidney Disease 1 Like 1 Gene Variants in Children With Biliary Atresia Splenic Malformation Syndrome // Hepatology. 2019. Т. 70, № 3. С. 899–910.
Gupta V. и др. Arthrogryposis, renal dysfunction and cholestasis (ARC) syndrome: a rare association with high GGT level and absent kidney // BMJ Case Rep. 2018. С. bcr-2017-223715.
Teker Düztaş D. и др. Two Cases With Neonatal Cholestasis and Renal Disorders Due to DCDC2 Mutation // Experimental and Clinical Transplantation. 2022. Т. 20, № Suppl 3. С. 115–117.
Пыков М.И. и др. Ультразвуковая диагностика изменений почек у детей с синдромом Алажилля. // Medical Council. Remedium, Ltd., 2017. № 9. С. 148–153.
Fattouh A.M. и др. The prevalence of congenital heart defects in infants with cholestatic disorders of infancy: a single-centre study // Arch Dis Child. 2016. Т. 101, № 9.
Волынец Г.В. и др. Дифференциальная диагностика и принципы терапии врожденных холестатических болезней у детей раннего возраста. 2018. 83 с.
Chitayat D., Kamath B., Saleh M. Alagille syndrome: clinical perspectives // Appl Clin Genet. 2016. Т. Volume 9.
Elisofon S.A. и др. Society of pediatric liver transplantation: Current registry status 2011‐2018 // Pediatr Transplant. 2020. Т. 24, № 1.
Liu Y. и др. Liver Transplantation for Progressive Familial Intrahepatic Cholestasis // Ann Transplant. 2018. Т. 23.
Jeyaraj R. и др. The Genetics of Inherited Cholestatic Disorders in Neonates and Infants: Evolving Challenges // Genes (Basel). 2021. Т. 12, № 11. С. 1837.
Sokol R.J. Fat-soluble vitamins and their importance in patients with cholestatic liver diseases // Gastroenterol Clin North Am. . 1994. Т. 23, № 4. С. 673–705.
Argao E.A., Heubi J.E. Fat-soluble vitamin deficiency in infants and children // Curr Opin Pediatr. 1993. Т. 5, № 5.
Shen Y.-M. и др. Oral Absorbable Fat-soluble Vitamin Formulation in Pediatric Patients With Cholestasis // J Pediatr Gastroenterol Nutr. 2012. Т. 55, № 5.
Feranchak A.P. и др. Comparison of indices of vitamin A status in children with chronic liver disease // Hepatology. 2005. Т. 42, № 4.
Feranchak A.P., Sokol R.J. Medical and Nutritional Management of Cholestasis in Infants and Children // Liver Disease in Children. Cambridge University Press, 2007.
Rovner A.J. и др. Rethinking Growth Failure in Alagille Syndrome: The Role of Dietary Intake and Steatorrhea // J Pediatr Gastroenterol Nutr. 2002. Т. 35, № 4.
Huang L. и др. Efficacy and safety of ursodeoxycholic acid in children with cholestasis: A systematic review and meta-analysis // PLoS One. 2023. Т. 18, № 1.
Moisseiev E., Cohen S., Dotan G. Alagille Syndrome Associated with Xerophthalmia // Case Rep Ophthalmol. 2013. Т. 4, № 3.
Bhatia V. и др. Management of neonatal cholestasis: Consensus statement of the pediatric gastroenterology chapter of Indian academy of pediatrics // Indian Pediatr. 2014. Т. 51, № 3. С. 203–210.
Hossain Z. и др. Effects of total enteral nutrition on early growth, immunity, and neuronal development of preterm infants // Nutrients. MDPI, 2021. Т. 13, № 8.
Tessitore M. и др. Malnutrition in pediatric chronic cholestatic disease: An up-to-date overview // Nutrients. MDPI, 2021. Т. 13, № 8.
Degrassi I. и др. Fat-Soluble Vitamins Deficiency in Pediatric Cholestasis: A Scoping Review // Nutrients. Multidisciplinary Digital Publishing Institute (MDPI), 2023. Т. 15, № 11.
Shneider B.L. и др. Efficacy of fat-soluble vitamin supplementation in infants with biliary atresia // Pediatrics. 2012. Т. 130, № 3.
Heinz N., Vittorio J. Treatment of Cholestasis in Infants and Young Children // Current Gastroenterology Reports. Springer, 2023. Т. 25, № 11. С. 344–354.
Dani C. и др. Italian guidelines for the management and treatment of neonatal cholestasis // Italian Journal of Pediatrics. BioMed Central Ltd., 2015. Т. 41, № 1.
Catzola A., Vajro P. Management options for cholestatic liver disease in children // Expert Review of Gastroenterology and Hepatology. Taylor and Francis Ltd, 2017. Т. 11, № 11. С. 1019–1030.
Chen R. и др. Clinical and genetic characterization of pediatric patients with progressive familial intrahepatic cholestasis type 3 (PFIC3): identification of 14 novel ABCB4 variants and review of the literatures // Orphanet J Rare Dis. 2022. Т. 17, № 1.
Ткаченко А.К. и др. Желтухи Неонатального Периода. Минск, 2017. Т. 68.
Rabbani T., Guthery S.L., Himes R., Shneider B.L., Harpavat S.. Newborn Screening for Biliary Atresia: a Review of Current Methods // Curr Gastroenterol Rep. 2021;23(12):28.
Rabbani T., Shah J. Newborn screening for biliary atresia using direct bilirubin: An implementation science study // J Med Screen. 2025;32(2):61-66.
Н.Л. Аряев, Н.В. Котова Неонатальные гепатиты Журнал ЗТД Журнал ЗТД №2 (38) 2013
Синдром желтухи у детей, критерии потенциальной опасности: учеб. пособие / Л.А. Мусатова, Л.И. Краснова, Н.С. Карташева [и др.]. – Пенза : Изд-во ПГУ, 2022. – 88 с.