Galanello R., Origa R. Beta-thalassemia. // Orphanet J Rare Dis. Orphanet J Rare Dis, 2010. Vol. 5. P. 11.
Steinberg M.H. Disorders of hemoglobin: genetics, pathophysiology, and clinical management. Cambridge University Press, 2001. 1268 p.
David J. Weatherall, J. B. Clegg. The Thalassaemia Syndromes, 4th Edition. 4th ed. John Wiley & Sons Limited, 2008.
Fucharoen S., Viprakasit V. Hb H disease: clinical course and disease modifiers // Hematology Am Soc Hematol Educ Program. Hematology Am Soc Hematol Educ Program, 2009. P. 26–34.
Weatherall D.J. The definition and epidemiology of non-transfusion-dependent thalassemia // Blood Rev. Blood Rev, 2012. Vol. 26 Suppl 1, № SUPPL.1.
Weatherall D.J. The inherited diseases of hemoglobin are an emerging global health burden // Blood. Blood, 2010. Vol. 115, № 22. P. 4331–4336.
Christianson A., Howson C.P., Modell B. March of dimes global report on birth defects.
Modell B., Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators // Bull World Health Organ. Bull World Health Organ, 2008. Vol. 86, № 6. P. 480–487.
Colah R., Gorakshakar A., Nadkarni A. Global burden, distribution and prevention of β-thalassemias and hemoglobin E disorders // Expert Rev Hematol. Expert Rev Hematol, 2010. Vol. 3, № 1. P. 103–117.
Сметанина Н.С. Талассемии у детей в России. Расширенное заседание Экспертного Совета Комитета Государственной Думы по охране здоровья по редким (орфанным) заболеваниям [Electronic resource]. 2024. URL: https://nacgenetic.ru/rasshirennoe-zasedanie-ekspertnogo-soveta-po-redkim-orfannym-zabolevaniyam-gd-rf (accessed: 09.05.2024).
Sripichai O. et al. A scoring system for the classification of beta-thalassemia/Hb E disease severity // Am J Hematol. Am J Hematol, 2008. Vol. 83, № 6. P. 482–484.
Vichinsky, E., Levine, L., Bhatia, S., Bojanowski, J., Coates, T. and Foote, D. (2012) Standards of Care Guidelines for Thalassemia. Children’s Hospital and Research Center, Oakland. - References - Scientific Research Publishing [Electronic resource]. URL: https://www.scirp.org/reference/referencespapers?referenceid=2061526 (accessed: 09.05.2024).
Farmakis D. et al. 2021 Thalassaemia International Federation Guidelines for the Management of Transfusion-dependent Thalassemia // Hemasphere. Hemasphere, 2022. Vol. 6, № 8.
Guidelines for the Clinical Management of Thalassaemia [Internet] - PubMed [Electronic resource]. URL: https://pubmed.ncbi.nlm.nih.gov/24308075/ (accessed: 09.05.2024).
Galanello R., Origa R. Beta-thalassemia // Orphanet J Rare Dis. Orphanet J Rare Dis, 2010. Vol. 5, № 1.
Ebrahimi M., Mohammadi-Asl J., Rahim F. The worldwide molecular spectrum and distribution of thalassaemia: a systematic review // Ann Hum Biol. Ann Hum Biol, 2021. Vol. 48, № 4. P. 307–312.
Bazazzadegan N. et al. The Spectrum of HBB Mutations among 2315 Beta Thalassemia Patients of a Reference Clinic in Tehran-Iran // Hemoglobin. Hemoglobin, 2023. Vol. 47, № 4. P. 147–151.
Henderson S.J. et al. Ten Years of Routine α- and β-Globin Gene Sequencing in UK Hemoglobinopathy Referrals Reveals 60 Novel Mutations // Hemoglobin. Hemoglobin, 2016. Vol. 40, № 2. P. 75–84.
Da Z.Z. et al. [Rare thalassemia caused by novel nucleotide variants in the globin gene: four case reports and literature review] // Zhonghua Xue Ye Xue Za Zhi. Zhonghua Xue Ye Xue Za Zhi, 2021. Vol. 42, № 4. P. 313–317.
Yang Z. et al. Gene spectrum analysis of thalassemia for people residing in northern China // BMC Med Genet. BMC Med Genet, 2019. Vol. 20, № 1.
Dong-Ming L.I., Sheng H.E. Genotypes of thalassemia in children: an analysis of 30 417 cases // Zhongguo Dang Dai Er Ke Za Zhi. Zhongguo Dang Dai Er Ke Za Zhi, 2021. Vol. 23, № 8. P. 841–847.
Hosseini-Bensenjan M. et al. Correlation of Serum Ferritin Level with Heart T2 MRI in Transfusion Dependent Thalassemia: a Systematic Review and Meta-Analysis // Clin Lab. Clin Lab, 2023. Vol. 69, № 6. P. 1063–1070.
Sarigianni M. et al. Accuracy of magnetic resonance imaging in diagnosis of liver iron overload: a systematic review and meta-analysis // Clin Gastroenterol Hepatol. Clin Gastroenterol Hepatol, 2015. Vol. 13, № 1. P. 55-63.e5.
Cappellini M.D. et al. A paradigm shift on beta-thalassaemia treatment: How will we manage this old disease with new therapies? // Blood Rev. Blood Rev, 2018. Vol. 32, № 4. P. 300–311.
Carrier Screening Programs for Cystic Fibrosis, Fragile X Syndrome, Hemoglobinopathies and Thalassemia, and Spinal Muscular Atrophy: A Health Technology Assessment - PubMed [Electronic resource]. URL: https://pubmed.ncbi.nlm.nih.gov/37637488/ (accessed: 09.05.2024).
Saffi M., Howard N. Exploring the Effectiveness of Mandatory Premarital Screening and Genetic Counselling Programmes for β-Thalassaemia in the Middle East: A Scoping Review // Public Health Genomics. Public Health Genomics, 2015. Vol. 18, № 4. P. 193–203.
Zafari M. et al. Non-invasive prenatal diagnosis of β-thalassemia by detection of the cell-free fetal DNA in maternal circulation: a systematic review and meta-analysis // Ann Hematol. Ann Hematol, 2016. Vol. 95, № 8. P. 1341–1350.
Suwannakhon N. et al. Noninvasive prenatal screening test for compound heterozygous beta thalassemia using an amplification refractory mutation system real-time polymerase chain reaction technique // Hematol Rep. Page Press Publications, 2019. Vol. 11, № 3. P. 65–69.
Rostamian H. et al. Prevalence and specificity of red blood cell alloantibodies and autoantibodies in transfused Iranian β-thalassemia patients: A systematic review and meta-analysis // Asian J Transfus Sci. Wolters Kluwer Medknow Publications, 2022. Vol. 16, № 1. P. 111–120.
Zafari M. et al. Non-invasive prenatal diagnosis of β-thalassemia by detection of the cell-free fetal DNA in maternal circulation: a systematic review and meta-analysis // Ann Hematol. Ann Hematol, 2016. Vol. 95, № 8. P. 1341–1350.
Suwannakhon N. et al. Non-invasive prenatal screening & diagnosis of ß-thalassaemia in an affected foetus // Indian Journal of Medical Research. Wolters Kluwer Medknow Publications, 2023. Vol. 157, № 5. P. 447–452.
Non-invasive prenatal diagnosis of β-thalassemia by detection of the cell-free fetal DNA in maternal circulation: a systematic review and meta-analysis - PubMed [Electronic resource]. URL: https://pubmed.ncbi.nlm.nih.gov/26968552/ (accessed: 09.05.2024).
Weatherall D.J. The inherited diseases of hemoglobin are an emerging global health burden // Blood. American Society of Hematology, 2010. Vol. 115, № 22. P. 4331–4336.
Hemoglobinopathies worldwide: present and future - PubMed [Electronic resource]. URL: https://pubmed.ncbi.nlm.nih.gov/18991645/ (accessed: 09.05.2024).
Weatherall D.J. The global problem of genetic disease // Ann Hum Biol. 2005. Vol. 32, № 2. P. 117–122.
Weatherall D. Hemoglobinopathies worldwide: present and future // Curr Mol Med. Curr Mol Med, 2008. Vol. 8, № 7. P. 592–599.
Weatherall D.J. The challenge of thalassemia for the developing countries // Ann N Y Acad Sci. New York Academy of Sciences, 2005. Vol. 1054. P. 11–17.
Weatherall D.J. The challenge of haemoglobinopathies in resource-poor countries // Br J Haematol. 2011. Vol. 154, № 6. P. 736–744.
Inherited haemoglobin disorders: an increasing global health problem - PubMed [Electronic resource]. URL: https://pubmed.ncbi.nlm.nih.gov/11545326/ (accessed: 09.05.2024).
Weatherall D.J. The global problem of genetic disease // Ann Hum Biol. 2005. Vol. 32, № 2. P. 117–122.
Weatherall D.J. The inherited diseases of hemoglobin are an emerging global health burden // Blood. Blood, 2010. Vol. 115, № 22. P. 4331–4336.
Shah F. et al. Relationship between Serum Ferritin and Outcomes in β-Thalassemia: A Systematic Literature Review // J Clin Med. J Clin Med, 2022. Vol. 11, № 15.
Goldberg E.K., Lal A., Fung E.B. Nutrition in Thalassemia: A Systematic Review of Deficiency, Relations to Morbidity, and Supplementation Recommendations // J Pediatr Hematol Oncol. J Pediatr Hematol Oncol, 2022. Vol. 44, № 1. P. 1–11.
Swe K.M.M. et al. Zinc supplements for treating thalassaemia and sickle cell disease // Cochrane Database Syst Rev. Cochrane Database Syst Rev, 2013. Vol. 2013, № 6.
Arab-Zozani M. et al. A Systematic Review and Meta-Analysis of Stature Growth Complications in β-thalassemia Major Patients // Ann Glob Health. Ann Glob Health, 2021. Vol. 87, № 1.
Henriksen L.F. et al. Increased iron stores prolong the QT interval - a general population study including 20 261 individuals and meta-analysis of thalassaemia major // Br J Haematol. Br J Haematol, 2016. Vol. 174, № 5. P. 776–785.
Patsourakos D. et al. Speckle tracking echocardiography and β-thalassemia major. A systematic review // Ann Hematol. Ann Hematol, 2023.
Haghpanah S. et al. The Prevalence of Hypothyroidism among Patients With β-Thalassemia: A Systematic Review and Meta-Analysis of Cross-Sectional Studies // Hemoglobin. Hemoglobin, 2021. Vol. 45, № 5. P. 275–286.
He L.N. et al. Elevated Prevalence of Abnormal Glucose Metabolism and Other Endocrine Disorders in Patients with β-Thalassemia Major: A Meta-Analysis // Biomed Res Int. Biomed Res Int, 2019. Vol. 2019.
Waheed U. et al. A Systematic Review and Meta-Analysis on the Epidemiology of Hepatitis B and Hepatitis C Virus among Beta-Thalassemia Major Patients in Pakistan // J Lab Physicians. J Lab Physicians, 2021. Vol. 13, № 3. P. 270–276.
Ehsan H. et al. Prevalence of Transfusion Transmissible Infections in Beta-Thalassemia Major Patients in Pakistan: A Systematic Review // Cureus. Cureus, 2020. Vol. 12, № 8.
Akhtar S., Nasir J.A., Hinde A. The prevalence of hepatitis C virus infection in β-thalassemia patients in Pakistan: a systematic review and meta-analysis // BMC Public Health. BMC Public Health, 2020. Vol. 20, № 1.
Behzadifar M., Gorji H.A., Bragazzi N.L. The prevalence of hepatitis C virus infection in thalassemia patients in Iran from 2000 to 2017: a systematic review and meta-analysis // Arch Virol. Arch Virol, 2018. Vol. 163, № 5. P. 1131–1140.
Dede A.D. et al. Thalassemia-associated osteoporosis: a systematic review on treatment and brief overview of the disease // Osteoporos Int. Osteoporos Int, 2016. Vol. 27, № 12. P. 3409–3425.
Charoenngam N., Rittiphairoj T., Ponvilawan B. Fracture prevalence in thalassemia: a systematic review and meta-analysis // Arch Osteoporos. Arch Osteoporos, 2021. Vol. 16, № 1.
Moosazadeh M. et al. Comparison of decayed, missing, filled teeth index between thalassemia major patients and control group in Iran: a systematic review and meta-analysis // BDJ Open. BDJ Open, 2020. Vol. 6, № 1.
Akcalı A. et al. Periodontal condition of patients with Thalassemia Major: A systematic review and meta-analysis // Arch Oral Biol. Arch Oral Biol, 2019. Vol. 102. P. 113–121.
Liaska A. et al. β-Thalassemia and ocular implications: a systematic review // BMC Ophthalmol. BMC Ophthalmol, 2016. Vol. 16, № 1.
RUDD C., EVANS P.J., PEENEY A.L. Ocular complications in thalassaemia minor // Br J Ophthalmol. Br J Ophthalmol, 1953. Vol. 37, № 6. P. 353–358.
Daneshmend T.K. Ocular findings in a case of haemoglobin H disease // Br J Ophthalmol. Br J Ophthalmol, 1979. Vol. 63, № 12. P. 842–844.
Magli A. et al. Ocular manifestations in thalassemia minor // Ophthalmologica. Ophthalmologica, 1982. Vol. 184, № 3. P. 139–146.
Badfar G. et al. Hearing loss in Iranian thalassemia major patients treated with deferoxamine: A systematic review and meta-analysis // Caspian J Intern Med. Caspian J Intern Med, 2017. Vol. 8, № 4. P. 239–249.
Badfar G. et al. Hearing loss in Iranian thalassemia major patients treated with deferoxamine: A systematic review and meta-analysis // Caspian J Intern Med. Babol University of Medical Sciences, 2017. Vol. 8, № 4. P. 239–249.
Demosthenous C. et al. Beta-thalassemia: renal complications and mechanisms: a narrative review // Hematology. Hematology, 2019. Vol. 24, № 1. P. 426–438.
Romadhon P.Z. et al. Markers of Renal Complications in Beta Thalassemia Patients with Iron Overload Receiving Chelation Agent Therapy: A Systematic Review // J Blood Med. J Blood Med, 2022. Vol. 13. P. 725–738.
Scoglio M. et al. Kidney Tubular Damage Secondary to Deferasirox: Systematic Literature Review // Children (Basel). Children (Basel), 2021. Vol. 8, № 12.
Arian M. et al. Biochemical Markers of Early Renal Dysfunction in Patients with β-thalassemia Major: A Systematic Review and Meta-analysis // Curr Med Chem. Curr Med Chem, 2023. Vol. 31.
Evangeli M., Mughal K., Porter J.B. Which psychosocial factors are related to chelation adherence in thalassemia? A systematic review // Hemoglobin. Hemoglobin, 2010. Vol. 34, № 3. P. 305–321.
Arian M. et al. Health-related quality of life (HRQoL) in beta-thalassemia major (β-TM) patients assessed by 36-item short form health survey (SF-36): a meta-analysis // Qual Life Res. Qual Life Res, 2019. Vol. 28, № 2. P. 321–334.
Reddy P.S., Locke M., Badawy S.M. A systematic review of adherence to iron chelation therapy among children and adolescents with thalassemia // Ann Med. Ann Med, 2022. Vol. 54, № 1. P. 326–342.
Lee W.J. et al. The impact of chelation compliance in health outcome and health related quality of life in thalassaemia patients: a systematic review // Health Qual Life Outcomes. Health Qual Life Outcomes, 2024. Vol. 22, № 1.
Guidelines for the Management of Non-Transfusion Dependent Thalassaemia (NTDT) [Internet] - PubMed [Electronic resource]. URL: https://pubmed.ncbi.nlm.nih.gov/24672826/ (accessed: 09.05.2024).
Lal A. Assessment and treatment of pain in thalassemia // Ann N Y Acad Sci. Ann N Y Acad Sci, 2016. Vol. 1368, № 1. P. 65–72.
Thalassaemia - Living with - NHS [Electronic resource]. URL: https://www.nhs.uk/conditions/thalassaemia/living-with/ (accessed: 09.05.2024).
Molazem Z. et al. The Effects of Nutrition, Exercise, and a Praying Program on Reducing Iron Overload in Patients With Beta-Thalassemia Major: A Randomized Clinical Trial // Iran J Pediatr. Iran J Pediatr, 2016. Vol. 26, № 5.
Soleiman Ekhtiari Y. et al. The Effect of an Intervention Based on the PRECEDE- PROCEED Model on Preventive Behaviors of Domestic Violence Among Iranian High School Girls. // Iran Red Crescent Med J. Iranian Red Crescent Society, 2013. Vol. 15, № 1. P. 21–28.
Maheri A. et al. Associations between a health-promoting lifestyle and quality of life among adults with beta-thalassemia major // Epidemiol Health. Epidemiol Health, 2016. Vol. 38. P. e2016050.
Fung E.B. et al. Relationships among Physical Activity, Pain, and Bone Health in Youth and Adults with Thalassemia: An Observational Study // Thalassemia reports. Thalass Rep, 2022. Vol. 12, № 3. P. 90–100.
Agostoni P. et al. Exercise capacity in patients with beta-thalassaemia intermedia // Br J Haematol. Br J Haematol, 2005. Vol. 131, № 2. P. 278–281.
Dehkordi A. et al. Effects of Aquatic Exercise on Dimensions of Quality of Life and Blood Indicators in Patients with Beta-Thalassemia Major // Int J Prev Med. Int J Prev Med, 2020. Vol. 11, № 1. P. 128.
Nanas S. et al. New insights into the exercise intolerance of beta-thalassemia major patients // Scand J Med Sci Sports. Scand J Med Sci Sports, 2009. Vol. 19, № 1. P. 96–102.
Vamvakas E.C., Blajchman M.A. Transfusion-related immunomodulation (TRIM): An update // Blood Rev. 2007. Vol. 21, № 6. P. 327–348.
Blajchman M.A. Immunomodulatory effects of allogeneic blood transfusions: Clinical manifestations and mechanisms // Vox Sang. Blackwell Publishing Ltd, 1998. Vol. 74, № SUPPL. 2. P. 315–319.
Vamvakas E.C. Pneumonia as a complication of blood product transfusion in the critically ill: transfusion-related immunomodulation (TRIM). // Crit Care Med. Lippincott Williams and Wilkins, 2006. Vol. 34, № 5 Suppl. P. S151-9.
Refaai M.A., Blumberg N. Transfusion immunomodulation from a clinical perspective: An update // Expert Rev Hematol. 2013. Vol. 6, № 6. P. 653–663.
Blajchman M.A. Transfusion immunomodulation or TRIM: what does it mean clinically? // Hematology. Hematology, 2005. Vol. 10 Suppl 1, № SUPPL. 1. P. 208–214.
F. Lindholm P., Annen K., Ramsey G. Approaches to minimize infection risk in blood banking and transfusion practice // Infect Disord Drug Targets. Infect Disord Drug Targets, 2011. Vol. 11, № 1. P. 45–56.
Sirchia G. et al. The clinical importance of leukocyte depletion in regular erythrocyte transfusions // Vox Sang. Vox Sang, 1986. Vol. 51 Suppl 1. P. 2–8.
Ozment C.P., Turi J.L. Iron overload following red blood cell transfusion and its impact on disease severity // Biochim Biophys Acta. Biochim Biophys Acta, 2009. Vol. 1790, № 7. P. 694–701.
Marwah S.S. et al. Increased non-transferrin bound iron in plasma-depleted SAG-M red blood cell units // Vox Sang. Vox Sang, 2002. Vol. 82, № 3. P. 122–126.
Chapman J.F. et al. Guidelines for pre-transfusion compatibility procedures in blood transfusion laboratories // Transfusion Medicine. Blackwell Publishing Ltd, 1996. Vol. 6, № 3. P. 273–283.
Milkins C. et al. Guidelines for pre-transfusion compatibility procedures in blood transfusion laboratories // Transfusion Medicine. John Wiley and Sons Inc, 2013. Vol. 23, № 1. P. 3–35.
Klein H.G., Spahn D.R., Carson J.L. Red blood cell transfusion in clinical practice // Lancet. Lancet, 2007. Vol. 370, № 9585. P. 415–426.
F. Lindholm P., Annen K., Ramsey G. Approaches to minimize infection risk in blood banking and transfusion practice // Infect Disord Drug Targets. Infect Disord Drug Targets, 2011. Vol. 11, № 1. P. 45–56.
Blajchman M.A. Transfusion immunomodulation or TRIM: what does it mean clinically? // Hematology. Hematology, 2005. Vol. 10 Suppl 1, № SUPPL. 1. P. 208–214.
Olivieri N.F. The beta-thalassemias // N Engl J Med. N Engl J Med, 1999. Vol. 341, № 2. P. 99–109.
Aydinok Y. Thalassemia // Hematology. Taylor & Francis, 2012. Vol. 17, № SUPPL. 1.
Lal A. et al. Combined chelation therapy with deferasirox and deferoxamine in thalassemia // Blood Cells Mol Dis. Blood Cells Mol Dis, 2013. Vol. 50, № 2. P. 99–104.
Deugnier Y. et al. Improvement in liver pathology of patients with β-thalassemia treated with deferasirox for at least 3 years // Gastroenterology. Gastroenterology, 2011. Vol. 141, № 4.
Wood J.C. et al. Follow-up report on the 2-year cardiac data from a deferasirox monotherapy trial // Am J Hematol. Am J Hematol, 2010. Vol. 85, № 10. P. 818–819.
Waldmeier F. et al. Pharmacokinetics, metabolism, and disposition of deferasirox in beta-thalassemic patients with transfusion-dependent iron overload who are at pharmacokinetic steady state // Drug Metab Dispos. Drug Metab Dispos, 2010. Vol. 38, № 5. P. 808–816.
Voskaridou E., Christoulas D., Terpos E. Successful chelation therapy with the combination of deferasirox and deferiprone in a patient with thalassaemia major and persisting severe iron overload after single-agent chelation therapies // Br J Haematol. Br J Haematol, 2011. Vol. 154, № 5. P. 654–656.
Bollig C. et al. Deferasirox for managing iron overload in people with thalassaemia // Cochrane Database Syst Rev. John Wiley and Sons, Inc. and the Cochrane Library, 2017. Vol. 2017, № 8.
Saleem A. et al. No difference in myocardial iron concentration and serum ferritin with deferasirox and deferiprone in pediatric patients with hemoglobinopathies: A systematic review and meta-analysis // Transfus Clin Biol. Transfus Clin Biol, 2023. Vol. 30, № 1. P. 69–74.
Taher A. et al. Efficacy and safety of deferasirox doses of >30 mg/kg per d in patients with transfusion-dependent anaemia and iron overload // Br J Haematol. Br J Haematol, 2009. Vol. 147, № 5. P. 752–759.
Dou H. et al. Effectiveness and Safety of Deferasirox in Thalassemia with Iron Overload: A Meta-Analysis // Acta Haematol. Acta Haematol, 2019. Vol. 141, № 1. P. 32–42.
Cappellini M.D., Taher A.T. The use of luspatercept for thalassemia in adults // Blood Adv. Blood Adv, 2021. Vol. 5, № 1. P. 326–333.
Chen N. et al. Population Pharmacokinetics and Exposure-Response Relationship of Luspatercept, an Erythroid Maturation Agent, in Anemic Patients With β-Thalassemia // J Clin Pharmacol. J Clin Pharmacol, 2021. Vol. 61, № 1. P. 52–63.
Cappellini M.D. et al. A Phase 3 Trial of Luspatercept in Patients with Transfusion-Dependent β-Thalassemia // N Engl J Med. N Engl J Med, 2020. Vol. 382, № 13. P. 1219–1231.
Taher A.T. et al. Luspatercept for the treatment of anaemia in non-transfusion-dependent β-thalassaemia (BEYOND): a phase 2, randomised, double-blind, multicentre, placebo-controlled trial // Lancet Haematol. Lancet Haematol, 2022. Vol. 9, № 10. P. e733–e744.
Kattamis A., et al., Safety data from the dose-finding cohorts: a phase 2a study of luspatercept in pediatric patients with β-thalassemia (at https://library.ehaweb.org/eha/2024/eha2024-congress/419603/antonis.kattamis.safety.data.from.the.dose-finding.cohorts.a.phase.2a.study.of.html access was done 15.11.2024 ).
Modell B. Total management of thalassaemia major // Arch Dis Child. Arch Dis Child, 1977. Vol. 52, № 6. P. 489–500.
Al-Salem A.H., Al-Dabbous I., Bhamidibati P. The role of partial splenectomy in children with thalassemia // European Journal of Pediatric Surgery. Hippokrates Verlag GmbH, 1998. Vol. 8, № 6. P. 334–338.
Taher A.T. et al. Splenectomy and thrombosis: the case of thalassemia intermedia // J Thromb Haemost. J Thromb Haemost, 2010. Vol. 8, № 10. P. 2152–2158.
Aydinok Y. Overview of Current Chelation Practices // Thalassemia Reports. MDPI AG, 2011. Vol. 1, № 12. P. e10.
Aessopos A. et al. Cardiovascular effects of splenomegaly and splenectomy in beta-thalassemia // Ann Hematol. Ann Hematol, 2005. Vol. 84, № 6. P. 353–357.
Splenectomy for haematological disorders - PubMed [Electronic resource]. URL: https://pubmed.ncbi.nlm.nih.gov/24798604/ (accessed: 09.05.2024).
Badawy S.M. et al. A systematic review of quality of life in sickle cell disease and thalassemia after stem cell transplant or gene therapy // Blood Adv. Blood Adv, 2021. Vol. 5, № 2. P. 570–583.
Baronciani D. et al. Hemopoietic stem cell transplantation in thalassemia: a report from the European Society for Blood and Bone Marrow Transplantation Hemoglobinopathy Registry, 2000-2010 // Bone Marrow Transplant. Bone Marrow Transplant, 2016. Vol. 51, № 4. P. 536–541.
Angelucci E. et al. Phlebotomy to Reduce Iron Overload in Patients Cured of Thalassemia by Bone Marrow Transplantation // Blood. Content Repository Only!, 1997. Vol. 90, № 3. P. 994–998.
Lucarelli G. et al. Marrow transplantation in patients with thalassemia responsive to iron chelation therapy // N Engl J Med. N Engl J Med, 1993. Vol. 329, № 12. P. 840–844.
Lucarelli G. et al. Bone marrow transplantation in patients with thalassemia // N Engl J Med. N Engl J Med, 1990. Vol. 322, № 7. P. 417–421.
Bone marrow transplantation in thalassemia - PubMed [Electronic resource]. URL: https://pubmed.ncbi.nlm.nih.gov/8560287/ (accessed: 10.05.2024).
Andreani M. et al. Persistence of Mixed Chimerism in Patients Transplanted for the Treatment of Thalassemia // Blood. Content Repository Only!, 1996. Vol. 87, № 8. P. 3494–3499.
Ruggeri A. et al. Umbilical Cord Blood Transplantation for Children with Thalassemia and Sickle Cell Disease // Biology of Blood and Marrow Transplantation. Elsevier, 2011. Vol. 17, № 9. P. 1375–1382.
Kanathezhath B., Walters M.C. Umbilical cord blood transplantation for thalassemia major // Hematol Oncol Clin North Am. Elsevier, 2010. Vol. 24, № 6. P. 1165–1177.
Bhattacharya N., Hollands P. Placental Umbilical Cord Blood Transfusion in Transfusion-Dependent Beta Thalassemic Patients: A Communication // Stem Cells Transl Med. John Wiley & Sons, Ltd, 2018. Vol. 7, № S1. P. S9–S9.
Locatelli F. et al. Related umbilical cord blood transplantation in patients with thalassemia and sickle cell disease // Blood. American Society of Hematology, 2003. Vol. 101, № 6. P. 2137–2143.
Politis C. The psychosocial impact of chronic illness // Ann N Y Acad Sci. Ann N Y Acad Sci, 1998. Vol. 850. P. 349–354.
Jaafari Z. et al. Prevalence of Depression among Iranian Patients with Beta-Thalassemia Major: A Systematic Review and Meta-analysis // Iran J Med Sci. Iran J Med Sci, 2022. Vol. 47, № 1. P. 15–24.
Tartaglione I. et al. Brain functional impairment in beta-thalassaemia: the cognitive profile in Italian neurologically asymptomatic adult patients in comparison to the reported literature // Br J Haematol. Br J Haematol, 2019. Vol. 186, № 4. P. 592–607.
Betts M. et al. Systematic Literature Review of the Burden of Disease and Treatment for Transfusion-dependent β-Thalassemia // Clin Ther. Clin Ther, 2020. Vol. 42, № 2. P. 322-337.e2.
Fisher E. et al. Psychological therapies for the management of chronic and recurrent pain in children and adolescents // Cochrane Database Syst Rev. Cochrane Database Syst Rev, 2018. Vol. 9, № 9.
Aydinok Y. et al. Psychosocial implications of Thalassemia Major // Pediatr Int. Pediatr Int, 2005. Vol. 47, № 1. P. 84–89.
Anie K.A., Massaglia P. Psychological therapies for thalassaemia // Cochrane Database Syst Rev. Cochrane Database Syst Rev, 2014. Vol. 2014, № 3.
Bryant R., Walsh T. Transition of the chronically ill youth with hemoglobinopathy to adult health care: an integrative review of the literature // J Pediatr Health Care. J Pediatr Health Care, 2009. Vol. 23, № 1. P. 37–48.
Цейтлин Г.Я., Сидоренко Л.В., Володин Н.Н. Организация медицинской и психолого-социальной реабилитации детей и подростков с онкологическими и гематологическими заболеваниями // Российский журнал детской гематологии и онкологии. 2014; 3: 59-65.
Потапчук А.А., Терентьев Ф.В. Влияние занятий физической реабилитацией на качество жизни подростков, перенесших трансплантацию гемопоэтических стволовых клеток // Ученые записки университета им. П. Ф. Лесгафта. Федеральное государственное бюджетное образовательное учреждение высшего образования «Национальный государственный университет физической культуры, спорта и здоровья им. П.Ф. Лесгафта, Санкт-Петербург», 2019. № 4 (170): 266-269.
Potapchuk A.A., Terentyev F.V. Physical rehabilitation of children and adolescents with oncohematological pathology after hematopoietic stem cell transplantation // The Scientific Notes of the Pavlov University. FSBEI HE I.P. Pavlov SPbSMU MOH Russia, 2022. Vol. 28, № 4. P. 56–62.
Скворцова Ю.В., Масчан А.А., Делягин В.М., Сидоренко Л.В., Цейтлин Г.Я., Володин Н.Н., Румянцев А.Г. Актуальные вопросы наблюдения, диагностики и реабилитации пациентов на отдаленных сроках после трансплантации гемопоэтических стволовых клеток // Российский журнал детской гематологии и онкологии. 2014; 2: 13-18.
Вольф С. Б. и др. Медицинская реабилитация и санаторно-курортное лечение. – ГрГМУ, 2017.
Епифанова А., … Е.А.-М.Г., 2015 undefined. Медицинская реабилитация // static.insales-cdn.com.
Королев А., Шевцов В. Медицинская реабилитация. 2019.
ГЭОТАР-Медиа–Москва Г.П.-Учебник., 2014 undefined. Медицинская реабилитация // e-library.sammu.uz.
Пономаренко Г.Н. Санаторно-курортное лечение // medkniga.com.ua
Разумов А. Н., Стародубов В. И., Пономаренко Г. Н. Санаторно-курортное лечение: национальное руководство //М.: ГЭОТАР-Медиа. – 2021. – С. 752.
Nairz M. et al. The struggle for iron - a metal at the host-pathogen interface // Cell Microbiol. Cell Microbiol, 2010. Vol. 12, № 12. P. 1691–1702.
Gordeuk V.R. et al. Iron status and the outcome of HIV infection: an overview // J Clin Virol. J Clin Virol, 2001. Vol. 20, № 3. P. 111–115.
Weinberg E.D. Iron availability and infection // Biochim Biophys Acta. Biochim Biophys Acta, 2009. Vol. 1790, № 7. P. 600–605.
Angelucci E. et al. Hepatic iron concentration and total body iron stores in thalassemia major // N Engl J Med. N Engl J Med, 2000. Vol. 343, № 5. P. 327–331.
Ang A.L. et al. Deferiprone Is Associated with Lower Serum Ferritin (SF) Relative to Liver Iron Concentration (LIC) Than Deferoxamine and Deferasirox- Implications for Clinical Practice // Blood. American Society of Hematology, 2010. Vol. 116, № 21. P. 4246–4246.
Garbowski M.W. et al. Calibration of Improved T2* Method for the Estimation of Liver Iron Concentration in Transfusional Iron Overload. // Blood. American Society of Hematology, 2009. Vol. 114, № 22. P. 2004–2004.
Kolnagou A. et al. Liver iron and serum ferritin levels are misleading for estimating cardiac, pancreatic, splenic and total body iron load in thalassemia patients: factors influencing the heterogenic distribution of excess storage iron in organs as identified by MRI T2* // Toxicol Mech Methods. Toxicol Mech Methods, 2013. Vol. 23, № 1. P. 48–56.
Koohi F., Kazemi T., Miri-Moghaddam E. Cardiac complications and iron overload in beta thalassemia major patients-a systematic review and meta-analysis // Ann Hematol. Ann Hematol, 2019. Vol. 98, № 6. P. 1323–1331.
Davis B.A., Porter J.B. Long-term outcome of continuous 24-hour deferoxamine infusion via indwelling intravenous catheters in high-risk β-thalassemia // Blood. Content Repository Only!, 2000. Vol. 95, № 4. P. 1229–1236.
Carpenter J.P. et al. On T2* magnetic resonance and cardiac iron // Circulation. Circulation, 2011. Vol. 123, № 14. P. 1519–1528.
Anderson L.J. et al. Myocardial iron clearance during reversal of siderotic cardiomyopathy with intravenous desferrioxamine: a prospective study using T2* cardiovascular magnetic resonance // Br J Haematol. Br J Haematol, 2004. Vol. 127, № 3. P. 348–355.
Chouliaras G.L. et al. Cardiac magnetic resonance in transfusion dependent thalassaemia: assessment of iron load and relationship to left ventricular ejection fraction // Br J Haematol. Br J Haematol, 2010. Vol. 151, № 4. P. 397–401.
Modell B. et al. Improved survival of thalassaemia major in the UK and relation to T2* cardiovascular magnetic resonance // J Cardiovasc Magn Reson. J Cardiovasc Magn Reson, 2008. Vol. 10, № 1.
Rahav G. et al. Severe infections in thalassaemic patients: prevalence and predisposing factors // Br J Haematol. Br J Haematol, 2006. Vol. 133, № 6. P. 667–674.
An increased risk of hormonal disorders, primarily diabetes, in individuals with β -thalassemia major: a retrospective analysis - PubMed [Electronic resource]. URL: https://pubmed.ncbi.nlm.nih.gov/38096537/ (accessed: 09.05.2024).
Haghpanah S. et al. The Prevalence of Hypothyroidism among Patients With β-Thalassemia: A Systematic Review and Meta-Analysis of Cross-Sectional Studies // Hemoglobin. Hemoglobin, 2021. Vol. 45, № 5. P. 275–286.
Haghpanah S. et al. Cytokine Levels in Patients with β-Thalassemia Major and Healthy Individuals: a Systematic Review and Meta-Analysis // Clin Lab. Clin Lab, 2022. Vol. 68, № 11. P. 2301–2309.
Taher A, Musallam KM, Cappellini MD. Guidrlines for the management of non-transfusion-dependent b-thalassemia (3rd edition). TIF 2023 https://thalassaemia.org.cy/publications/tif-publications/guidelines-for-the-management-of-non-transfusion-dependent-%ce%b2-thalassaemia-3rd-edition-2023
Taher A, Farmakis D, Porter J, Cappellini MD, Musallam KM. Guidelines for the management of transfusion dependent thalassemia (5th edition – Version 2.0); TIF 2025 https://thalassaemia.org.cy/publications/tif-publications/guidelines-for-the-management-of-transfusion-dependent-%ce%b2-thalassaemia-5th-edition-2025/
Farmakis D. A short guide for the management of transfusion-dependent thalassemia (2nd edition – 2022), TIF https://thalassaemia.org.cy/publications/tif-publications/a-short-guide-for-the-management-of-transfusion-dependent-thalassaemia-2022
Aparna A Sagare, Dhiraj J Trivedi. Assesment of transferrin saturation as an indicator of iron overload in homozygous & hetrozygous form of thalassemia // Research Journal of Pharmaceutical Biological and Chemical Sciences, January 2014, 5(1):668-673
Chapchap EC, Silva MMA, Baroni RH, Araujo ADS, de Assis RA, Loggetto SR, Junior AF, Verissimo MPA, Baldanzi GR, Fertrin KY, Tricta F, Piga AG, Hamerschlak N. Extramedullary haematopoiesis in patients with thalassemia: a cross-sectional description of its prevalence, clinical features and survival. Hematol Transfus Cell Ther. 2024 Nov;46 Suppl 5(Suppl 5):S143-S151
YL Chan, HY Tse. Imaging in Thalassaemia. J HK Coll Radiol 2002;5:155-161.
Mangia, A., Bellini, D., Cillo, U. et al. Hepatocellular carcinoma in adult thalassemia patients: an expert opinion based on current evidence. BMC Gastroenterol 20, 251 (2020).
Isma'eel H, Arnaout MS, Shamseddeen W, Mahfouz R, Zeineh N, Jradi O, Taher A. Screening for inherited thrombophilia might be warranted among Eastern Mediterranean sickle-beta-0 thalassemia patients. J Thromb Thrombolysis. 2006 Oct;22(2):121-3.
Seregina EA, Nikulina OF, Tsvetaeva NV, Rodionova MN, Gribkova IV, Orel EB, Zapariy AP, Erasov AV, Balandina AN, Ananyeva NM, Ataullakhanov FI. Laboratory tests for coagulation system monitoring in a patient with β-thalassemia. Int J Hematol. 2014;99(5):588-96
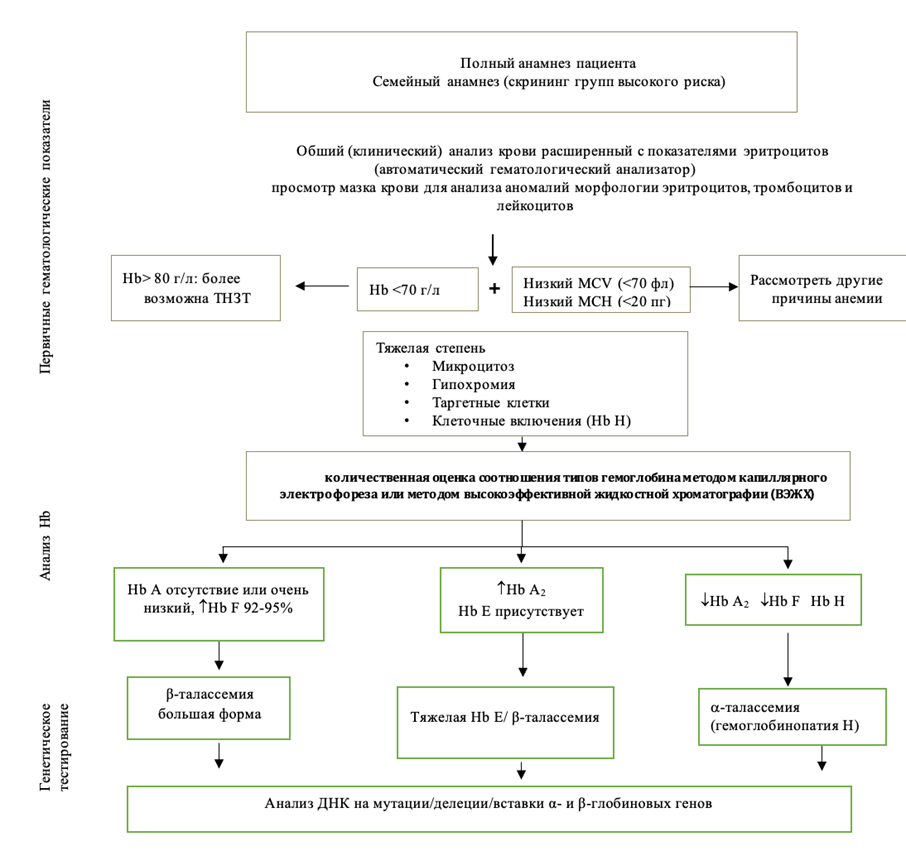
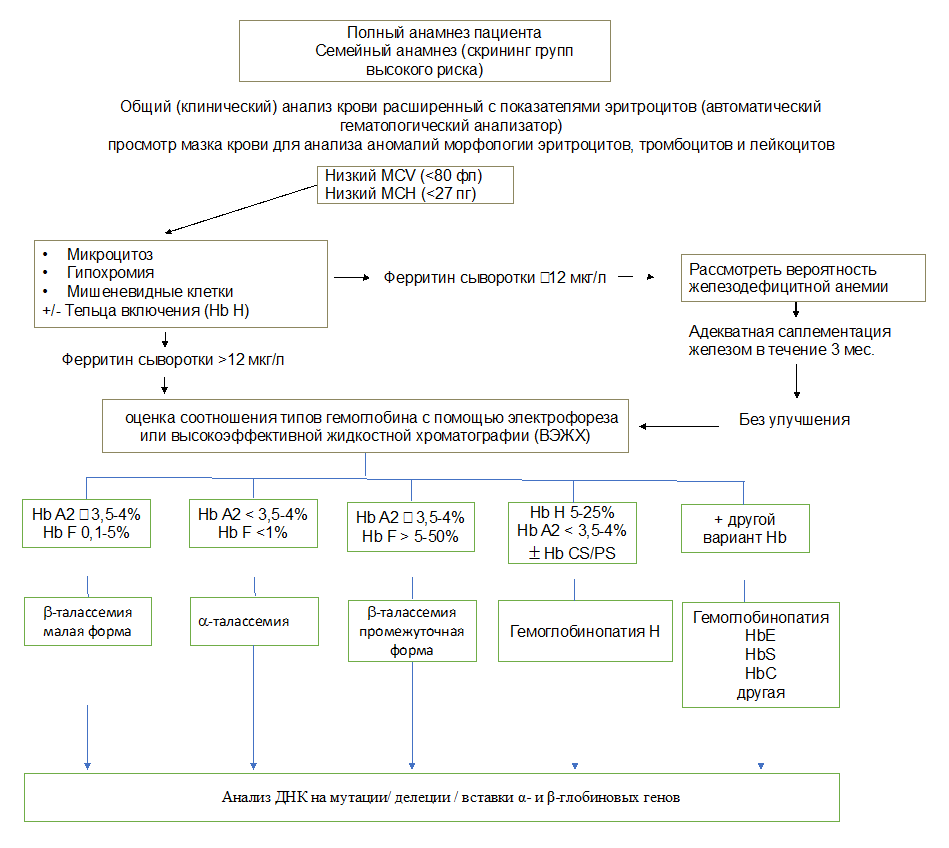
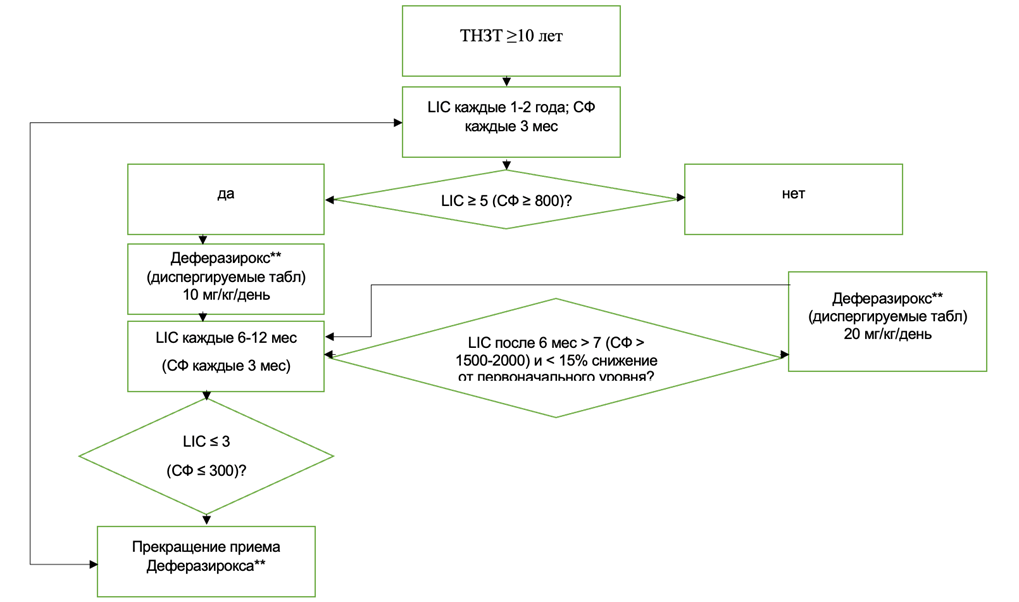 Б4. Лечебная тактика при ТНЗТ (например, промежуточной форме β-талассемии)
Б4. Лечебная тактика при ТНЗТ (например, промежуточной форме β-талассемии)