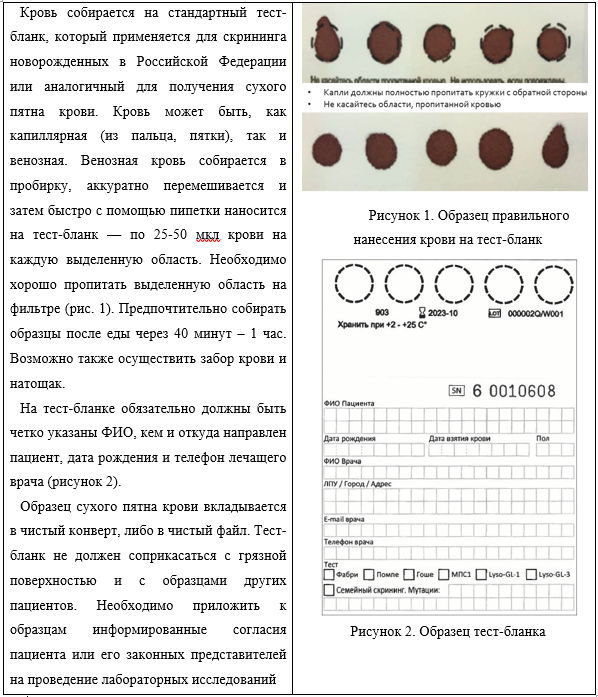
Germain D.P. Fabry disease//Orphanet J Rare Dis. 2010. V.22. P. 5-30.
Spada M., Pagliardini S. et al. High incidence of later-onset Fabry disease revealed by newborn screening// Am J Hum Genet. 2006. V.79. P. 31-40.
Hwu W.L., Chien Y.H. et al. Newborn screening for Fabry disease in Taiwan reveals a high incidence of the later-onset GLA mutation c.936+919G > A (IVS4+919G > A)// Hum Mutat. 2009. V.30. P.1397-1405.
Elstein D., Altarescu Gh. et al. Fabry disease// Springer Science & Business Media. 2010.
Hopkin R.J., Bissler J. et al. Characterization of Fabry Disease in 352 Pediatric Patients in the Fabry Registry// Pediatr Res. 2008. V. 64. P. 550-5.
Hoffmann B., Beck M. et al. Nature and prevalence of pain in Fabry disease and its response to enzyme replacement therapy–a retrospective analysis from the Fabry Outcome Survey// Clin J Pain. 2007. V.23. P.535-42.
Sims K., Politei J. et al. Stroke in Fabry disease frequently occurs before diagnosis and in the absence of other clinical events: natural history data from the Fabry Registry// Stroke. 2009. V.40. P.788-94.
Mehta A., Clarke J.T. et al. Natural course of Fabry disease: changing pattern of causes of death in FOS - Fabry Outcome Survey//J Med Genet. 2009.V.46. P.548-52.
Ryan P. Morrissey. Cardiac abnormalities in Anderson-Fabry disease and Fabry’s cardiomyopathy// Cardiovasc J Afr. 2011. V. 22. №1. P. 38–44.
Sakuraba H., Togawa T., Tsukimura T. et al. Plasma lyso-Gb3: a biomarker for monitoring Fabry patients during enzyme replacement therapy// Clin Exp Nephrol. 2018. V.22. №4. P. 843-9.
Schiffmann R., Warnock D.G. et al. Fabry disease: progression of nephropathy, and prevalence of cardiac and cerebrovascular events before enzyme replacement therapy// Nephrol Dial Transplant. 2009. V.24. №7. P. 2102–2111.
Linhart A., Kampmann C., Zamorano J.L. et al. Cardiac manifestations of Anderson-Fabry disease: results from the international Fabry outcome survey// Eur Heart J. 2007. V.28. P.1228–1235.
Weidemann F., Breunig F., Beer M. et al. The variation of morphological and functional cardiac manifestation in Fabry disease: potential implications for the time course of the disease// Eur Heart J. 2005. V. 26. P.1221–7.
Gupta S., Ries M. et al. The relationship of vascular glycolipid storage to clinical manifestations of Fabry disease: a cross-sectional study of a large cohort of clinically affected heterozygous women// Medicine (Baltimore). 2005. V.84. №5. P. 261-8.
Cordeiro C.A., Oréfice F. et al. Córnea verticilata - marcador clínico da doença de Fabry: relato de caso// Arq Bras Oftalmol. 2007. V.70. №4. P.701-5.
Mayes J.S., Scheerer J.B. et al. Differential assay for lysosomal alpha-galactosidases in human tissues and its application to Fabry’s disease// Clin Chim Acta. 1981.V. 112. P.247-251.
Linthorst G.E., Vedder A.C., Aerts J.M., Hollak C.E. Screening for Fabry disease using whole blood spots fails to identify one-third of female carriers// Clin Chim Acta. 2005. V.353. P.201-3.
Linthorst G.E., Bouwman M.G., Wijburg F.A.et al. Screening for Fabry disease in high-risk populations: a systematic review//Journal of Medical Genetics. 2010. V.47. P.217-22.
Keating G.M. Agalsidase alfa: a review of its use in the management of Fabry disease// BioDrugs. 2012. V.26. №5. P.335-54.
Keating G.M., Simpson D. Agalsidase Beta: a review of its use in the management of Fabry disease// Drugs. 2007. V.67. №3. P.435-55.
Schiffmann R. Agalsidase treatment for Fabry disease: uses and rivalries// Genet Med. 2010. V.12. №11. P.684-5.
Banikazemi M., Bultas J. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial//Ann Intern Med. 2007. V.146. №2. P.77-86.
Tøndel C., Bostad L. et al. Agalsidase benefits renal histology in young patients with Fabry disease// J Am Soc Nephrol. 2013. V.24. №1. P.137-48.
Warnock D.G., Ortiz A. et al. Renal outcomes of agalsidase beta treatment for Fabry disease: role of proteinuria and timing of treatment initiation// Nephrol Dial Transplant. 2012.V. 27. №3. P.1042-9.
Bénichou B., Goyal S. A retrospective analysis of the potential impact of IgG antibodies to agalsidase beta on efficacy during enzyme replacement therapy for Fabry disease// Mol Genet Metab. 2009. V.96. №1. P.4-12.
Ortiz A., Germain D.P., Desnick R.J. et al. Fabry disease revisited: Management and treatment recommendations for adult patients// Molecular Genetics and Metabolism. 2018. V.123. №4. P. 416-27.
Mehta A., West M.L. Therapeutic goals in the treatment of Fabry disease// Genet Med. 2010. V.12. №11. P.713-20..
Laney D.A., Bennett R.L., Clarke V. et al. Fabry disease practice guidelines: recommendations of the National Society of Genetic Counselors// J Genet Couns. 2013. V.22. №5. P.555-64.
Desnick R.J., Brady R., Barranger J. et al. Fabry disease, an under-recognized multisystemic disorder: expert recommendations for diagnosis, management, and enzyme replacement therapy// Ann Intern Med. 2003. V.138. №4. P.338-46.
Polo G., Burlina A., Ranieri E. et al. Plasma and dried blood spot lysosphingolipids for the diagnosis of different sphingolipidoses: a comparative study// Clinical Chemistry and Laboratory Medicine (CCLM). 2019. V.57. №12. P.1863-74.
Mariëlle J. van Breemen et al. Reduction of elevated plasma globotriaosylsphingosine in patients with classic Fabry disease following enzyme replacement therapy// Biochimica et Biophysica Acta. 2011.V.1812. P. 70–76.
Lenders M., Canaan-Kühl S. et al. Patients with Fabry Disease after Enzyme Replacement Therapy Dose Reduction and Switch–2-Year Follow-Up// J Am Soc Nephrol. 2016. V.27. P. 952–962.
Goker-Alpan O., Gambello M. J. et al. Reduction of Plasma Globotriaosylsphingosine Levels After Switching from Agalsidase Alfa to Agalsidase Beta as Enzyme Replacement Therapy for Fabry Disease// JIMD Rep. 2015.V.25. P. 95-106.
Beck M., Hughes D. et al. Long-term effectiveness of agalsidase alfa enzyme replacement in Fabry disease: A Fabry Outcome Survey analysis// Mol Genet Metab Rep. 2015. V.3. P.21-7.
Politei J.M., Bouhassira D., Germain D.P. et al. Pain in Fabry disease: practical recommendations for diagnosis and treatment// CNS Neuroscience and therapeutics. 2016. V.22. P. 568-576.
Shi Q., Chen J., Pongmoragot J. et al. Prevalence of Fabry disease in stroke patients – a systematic review and meta-analysis// J. Stroke cerebrovasc. Dis. 2014.V. 23. №5. P.985-92.
Fuller M. et al. Immunoquantification of α-galactosidase: evaluation for the diagnosis of Fabry disease //Clinical chemistry. 2004. V.50. №.11. P.1979-85.
Kramer J., Weidemann F. Biomarkers for diagnosing and staging of Fabry disease //Current medicinal chemistry. 2018. V. 25. №13. P.1530-7.
Rombach S. M. et al. The value of estimated GFR in comparison to measured GFR for the assessment of renal function in adult patients with Fabry disease //Nephrology Dialysis Transplantation. 2010. V. 25. № 8. P. 2549-56.
Körver S. et al. Development and clinical consequences of white matter lesions in Fabry disease: a systematic review //Molecular genetics and metabolism. 2018. V. 125. № 3. P. 205-216.
van den Boomen M. et al. Native T1 reference values for nonischemic cardiomyopathies and populations with increased cardiovascular risk: A systematic review and meta‐analysis //Journal of Magnetic Resonance Imaging. 2018. V. 47. № 4. P. 891-912.
Van der Tol L., Sminia M. L., Hollak C. E. M., Biegstraaten M. Cornea verticillata supports a diagnosis of Fabry disease in non-classical phenotypes: results from the Dutch cohort and a systematic review// British Journal of Ophthalmology.2015. V.100. №1. P. 3–8. doi:10.1136/bjophthalmol-2014-306433
Mohr J.P. Stroke: pathophysiology, diagnosis and management// Elsevier, sixth edition, 2016.
Alegra T., Vairo F., de Souza M.V. et al., Enzyme replacement therapy for Fabry disease: A systematic review and meta-analysis// Genet Mol Biol. 2012.V.35. P.947-54.
Spada M., Baron R., Elliott P.M. et al. The effect of enzyme replacement therapy on clinical outcomes in paediatric patients with Fabry disease - A systematic literature review by a European panel of experts// Mol Genet Metab. 2019. V.126. №3.P.212-23.
Germain D.P., Elliott P.M., Falissard B. et al. The effect of enzyme replacement therapy on clinical outcomes in male patients with Fabry disease: A systematic literature review by a European panel of experts// Mol Genet Metab Rep. 2019. V.19. P.100454.
Germain D.P., Arad M., Burlina A. et al. The effect of enzyme replacement therapy on clinical outcomes in female patients with Fabry disease - A systematic literature review by a European panel of experts// Mol Genet Metab. 2018. V.126. №3.P.224-35.
Biegstraaten M. et al. Recommendations for initiation and cessation of enzyme replacement therapy in patients with Fabry disease: the European Fabry Working Group consensus document// Orphanet J Rare Dis. 2015. V.10. P.36.
Ramaswami U., Whybra C., Parini R. et al. FOS European Investigators. Clinical manifestations of Fabry disease in children: data from the Fabry Outcome Survey//Acta Paediatr. 2006. V.95. №1. P.86–92.
El Dib R., Gomaa H., Ortiz A. et.al. Enzyme replacement therapy for Anderson-Fabry disease: A complementary overview of a Cochrane publication through a linear regression and a pooled analysis of proportions from cohort studies// PLoS ONE. 2017. V.12. №3. P. e0173358.
Schuller Y. et al. Pain management strategies for neuropathic pain in Fabry disease-a systematic review //BMC neurology. 2016. V.16. №1. P. 25.
Ersözlü S. et al. Long-Term Outcomes of Kidney Transplantation in Fabry Disease //Transplantation. 2018. V. 102. № 11. P. 1924-33.
Di L. Z. et al. Severe bradyarrhythmia linked to left atrial dysfunction in Fabry disease—A cross‐sectional study //Clinical cardiology. 2018. V. 41. №. 9. P. 1207-13.
Morais P. et al. Angiokeratomas of Fabry successfully treated with intense pulsed light //Journal of Cosmetic and Laser Therapy. 2008. V. 10. №4. P. 218-22.
Bolsover F. E. et al. Cognitive dysfunction and depression in Fabry disease: a systematic review //Journal of inherited metabolic disease. 2014. V. 37. № 2. P. 177-87.
Favalli V. et al. Genetic screening of Anderson-Fabry disease in probands referred from multispecialty clinics //Journal of the American College of Cardiology. 2016. V. 68. № 10. P. 1037-50.
Sirrs M. et al. Outcomes of patients treated through the Canadian Fabry disease initiative //Molecular genetics and metabolism. 2014. V. 111. № 4. P. 499-506.
Pieroni M. Поражение сердца при болезни Фабри: новые механизмы развития и подходы к лечению. Клин фармакол тер 2021;30(2):6-16 [Pieroni M. Cardiomyopathy in Fabry disease: insights in the pathogenesis and new treatment options. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2021;30(2):6-16 (In Russ.)]
Hagège A, Réant P, Habib G, et al. Fabry disease in cardiology practice: Literature review and expert point of view. Arch Cardiovasc Dis. 2019;112(4):278-287. doi:10.1016/j.acvd.2019.01.002
Hughes D.A., Ramaswam U., Elliott P. et. al. Guidelines for the Diagnosis and Management of Anderson-Fabry Disease. — 2008. — 32 p.
El-Abassi R, Singhal D, England JD. Fabry"s disease. J Neurol Sci. 2014;344(1-2):5-19. doi:10.1016/j.jns.2014.06.029
Волгина, С. Я. (2012). Болезнь Фабри. Практическая медицина, (7 (62)), 75-79.
Germain DP, Fouilhoux A, Decramer S, et al. Consensus recommendations for diagnosis, management and treatment of Fabry disease in paediatric patients. Clin Genet. 2019;96(2):107-117. doi:10.1111/cge.13546
Oliveira JP, Valbuena C, Baldaia Moreira A, et al. Splenomegaly, hypersplenism and peripheral blood cytopaenias in patients with classical Anderson-Fabry disease [published correction appears in Virchows Arch. 2011 Nov;459(5):555-6]. Virchows Arch. 2008;453(3):291-300. doi:10.1007/s00428-008-0651-4
Visser SL, de Groot WP. Electroencephalographic and electromyographic changes in a case of angiokeratoma corporis diffusum (Fabry"s disease). Confin Neurol. 1970;32(1):25-32. doi:10.1159/000103391
Ginsberg L. Nervous system manifestations of Fabry disease: data from FOS – the Fabry Outcome Survey. In: Mehta A, Beck M, Sunder-Plassmann G, eds. Fabry Disease: Perspectives from 5 Years of FOS. Oxford: Oxford PharmaGenesis; 2006.
Sudheera Magage, Jean-Claude Lubanda, Dominique P. Germain, Jan Bultas, Debora Karetová and Alès Linhart. Atteinte respiratoire de la maladie de Fabry. Med Sci (Paris), 21 (2005) 37-39. DOI: https://doi.org/10.1051/medsci/20052111s37
Vaisbich MH, Andrade LGM, Silva CAB, Barreto FC. Recommendations for the diagnosis and management of Fabry disease in pediatric patients: a document from the Rare Diseases Committee of the Brazilian Society of Nephrology (Comdora-SBN). J Bras Nefrol. 2022 Apr-Jun;44(2):268-280. doi: 10.1590/2175-8239-JBN-2021-0216
Моисеев А.С., Буланов Н.М., Тао Е.А. и др. Эффективность и безопасность длительной ферментозаместительной терапии агалсидазой альфа и агалсидазой бета у взрослых пациентов с болезнью Фабри. Клин фармакол тер 2022;31(4):28-34
Моисеев А.С., Мершина Е.А., Сафарова А.Ф. и др. Поражение сердца при болезни Фабри: особенности течения и диагностическое значение магнитно-резонансной томографии и speckle-tracking эхокардиографии. Клин фармакол тер 2022;31(3):22-29
Моисеев А.С., Тао Е.А., Буланов Н.М. и др. Поражение центральной нервной системы при болезни Фабри. Клин фармакол тер 2022;31(1): 32-38
Куцев С.И., Моисеев С.В. Семейный генетический скрининг при редких наследственных заболеваниях (на примере болезни Фабри). Клин фармакол тер 2021; 30(4):6-12
Моисеев С.В., Тао Е.А., Моисеев А.С. и др. Клинические проявления и исходы болезни Фабри у 150 взрослых пациентов. Клин фармакол тер 2021;30(3):43-51
Moiseev S, Tao E, Moiseev A, Bulanov N, Filatova E, Fomin V, Germain DP. The Benefits of Family Screening in Rare Diseases: Genetic Testing Reveals 165 New Cases of Fabry Disease among At-Risk Family Members of 83 Index Patients. Genes (Basel). 2022 Sep 9;13(9):1619
Moiseev S, Karovaikina E, Moiseev A, Bulanov N, Fomin V. Strategies of Screening for Fabry Disease in Patients with Unexplained Left Ventricular Hypertrophy. Mayo Clin Proc. 2019 Aug;94(8):1644-1646
Moiseev S, Fomin V, Savostyanov K, Pushkov A, Moiseev A, Svistunov A, Namazova-Baranova L. The Prevalence and Clinical Features of Fabry Disease in Hemodialysis Patients: Russian Nationwide Fabry Dialysis Screening Program. Nephron. 2019;141(4):249-255
Hwang S, Lee BH, Kim WS, Kim DS, Cheon CK, Lee CH, Choi Y, Choi JH, Kim JH, Yoo HW. A phase II, multicenter, open-label trial to evaluate the safety and efficacy of ISU303 (Agalsidase beta) in patients with Fabry disease. Medicine (Baltimore). 2022 Sep 16;101(37):e30345. doi: 10.1097/MD.0000000000030345
Cole AL, Lee PJ, Hughes DA, et al. Depression in adults with Fabry disease: a common and underdiagnosed problem. J Inherit Metab Dis. 2007; 30: 943-951
Wanner C., Arad M., Baron R. et al. European expert consensus statement on therapeutic goals in Fabry disease. Mol Genet Metab. 2018 Jul;124(3):189-203. doi: 10.1016/j.ymgme.2018.06.004
Nowicki M., Bazan-Socha S., Błażejewska-Hyzorek B. et al. Enzyme replacement therapy in Fabry disease in Poland: a position statement. Pol Arch Intern Med. 2020 Jan 31;130(1):91-97. doi: 10.20452/pamw.15117
Sirrs S, Bichet DG, Iwanochko RM, et al. Canadian Fabry disease treatment guidelines 2017. Toronto, Canada: The Garrod Association; 2017 Sep 22. http://www.garrod.ca/wp-content/uploads/Canadian-FD-Treatment-Guidelines-2017.pdf
Savostyanov K, Pushkov A, Zhanin I, Mazanova N, Trufanov S, Pakhomov A, Alexeeva A, Sladkov D, Asanov A, Fisenko A. The prevalence of Fabry disease among 1009 unrelated patients with hypertrophic cardiomyopathy: a Russian nationwide screening program using NGS technology. Orphanet J Rare Dis. 2022 May 16;17(1):199. doi: 10.1186/s13023-022-02319-4
Кузенкова Л.М., Намазова-Баранова Л.С., Подклетнова Т.В. и др. Болезнь Фабри: особенности заболевания у детей и подростков / Вопросы современной педиатрии. – 2015. – Т. 14. – № 3. – С. 341-348. – DOI 10.15690/vsp.v14i3.1369
Савостьянов К.В. Современные алгоритмы генетической диагностики редких наследственных болезней у российских пациентов. Научное издание / К.В. Савостьянов – Москва – Полиграфист и издатель – 2022 – 452 стр.
Mehta A, Beck M, Sunder-Plassmann G, editors Oxford: Oxford PharmaGenesis Chapter 27 Pulmonary involvement in Fabry disease; 2006 https://www.ncbi.nlm.nih.gov/books/NBK11589/
Приказ Минздрава России от 06.12.2021 №1122н «Об утверждении национального календаря профилактических прививок и календаря профилактических прививок по эпидемическим показаниям»
Методические указания МУ 3.3.1.1095—02. Медицинские противопоказания к проведению профилактических прививок препаратами национального календаря прививок
Laney DA, Germain DP, Oliveira JP, Burlina AP, Cabrera GH, Hong GR, Hopkin RJ, Niu DM, Thomas M, Trimarchi H, Wilcox WR, Politei JM, Ortiz A. Fabry disease and COVID-19: International expert recommendations for management based on real-world experience. Mol Genet Metab. 2021 Feb;132(2):S62
Tartour E, Germain DP. Humoral Immune Response to SARS-CoV-2 Vaccination after a Booster Vaccine Dose in Two Kidney Transplant Recipients with Fabry Disease and Variable Secondary Immunosuppressive Regimens. Vaccines (Basel). 2021 Nov 30;9(12):1412
Методические рекомендации по выявлению, расследованию и профилактике побочных проявлений после иммунизации. М., 2019. — 56 с.
Braga MC, Fonseca FLA, Marins MM, Gomes CP, Bacci MR, Martins AM, D"Almeida V. Evaluation of Beta 2-Microglobulin, Cystatin C, and Lipocalin-2 as Renal Biomarkers for Patients with Fabry Disease. Nephron. 2019;143(4):217-227
Muntean C, Starcea IM, Stoica C, Banescu C. Clinical Characteristics, Renal Involvement, and Therapeutic Options of Pediatric Patients With Fabry Disease. Front Pediatr. 2022 Jun 1;10:908657. doi: 10.3389/fped.2022.908657. Erratum in: Front Pediatr. 2022 Oct 19;10:1045199
Ezgu F, Alpsoy E, Bicik Bahcebasi Z, Kasapcopur O, Palamar M, Onay H, Ozdemir BH, Topcuoglu MA, Tufekcioglu O. Expert opinion on the recognition, diagnosis and management of children and adults with Fabry disease: a multidisciplinary Turkey perspective. Orphanet J Rare Dis. 2022 Mar 2;17(1):90.
Faverio P, Stainer A, De Giacomi F, Gasperini S, Motta S, Canonico F, Pieruzzi F, Monzani A, Pesci A, Biondi A. Molecular Pathways and Respiratory Involvement in Lysosomal Storage Diseases. Int J Mol Sci. 2019 Jan 15;20(2):327. doi: 10.3390/ijms20020327.
Svensson CK, Feldt-Rasmussen U, Backer V. Fabry disease, respiratory symptoms, and airway limitation - a systematic review. Eur Clin Respir J. 2015 Jun 26;2.
Yazdanfard PDW, Effraimidis G, Madsen CV, Nielsen LH, Rasmussen ÅK, Petersen JH, Sørensen SS, Køber L, Fraga de Abreu VH, Larsen VA, Feldt-Rasmussen U. Hearing loss in fabry disease: A 16 year follow-up study of the Danish nationwide cohort. Mol Genet Metab Rep. 2022 Feb 15;31:100841
Radulescu D, Crisan D, Militaru V, Buzdugan E, Stoicescu L, Grosu A, Vlad C, Grapa C, Radulescu ML. Gastrointestinal Manifestations and Treatment Options in Fabry Disease Patients. A Systematic Review. J Gastrointestin Liver Dis. 2022 Mar 19;31(1):98-106
Gugelmo G, Vitturi N, Francini-Pesenti F, Fasan I, Lenzini L, Valentini R, Carraro G, Avogaro A, Spinella P. Gastrointestinal Manifestations and Low-FODMAP Protocol in a Cohort of Fabry Disease Adult Patients. Nutrients. 2023 Jan 28;15(3):658
Effraimidis G, Rasmussen ÅK, Dunoe M, Hasholt LF, Wibrand F, Sorensen SS, Lund AM, Kober L, Bundgaard H, Yazdanfard PDW, Oturai P, Larsen VA, de Abreu VHF, Enevoldsen LH, Kristensen T, Svenstrup K, Bille MB, Arif F, Mogensen M, Klokker M, Backer V, Kistorp C, Feldt-Rasmussen U. Systematic cascade screening in the Danish Fabry Disease Centre: 20 years of a national single-centre experience. PLoS One. 2022 Nov 16;17(11):e0277767
Kouvaras SN. Endoscopic images in Fabry disease. Ann Gastroenterol. 2012;25(4):354. PMID: 24714151; PMCID: PMC3959418. Politei JM, Solar B. Gastrointestinal involvement in Fabry disease. Rare Disease and Orphan Drugs Journal. 2024; 3(2): 17. http://dx.doi.org/10.20517/rdodj.2023.46
Lux TJ, Meining A. Endoscopic treatment of a Fabry disease-related, lumen-obstructing colonic tumor. United European Gastroenterol J. 2023; 11(7): 690–691. https://doi.org/10.1002/ueg2.12418
Caputo, F.; Lungaro, L.; Galdi, A.; Zoli, E.; Giancola, F.; Caio, G.; De Giorgio, R.; Zoli, G. Gastrointestinal Involvement in Anderson-Fabry Disease: A Narrative Review. Int. J. Environ. Res. Public Health 2021, 18, 3320. https://doi.org/10.3390/ijerph18063320
Kurschat CE. Fabry disease-what cardiologists can learn from the nephrologist: a narrative review. Cardiovasc Diagn Ther. 2021 Apr;11(2):672-682. doi: 10.21037/cdt-20-981. PMID: 33968644; PMCID: PMC8102258.
By Muntean C, Starcea IM, Stoica C, Banescu C. Clinical characteristics, renal involvement, and therapeutic options of pediatric patients with Fabry disease (2022) Front Pediatr. 10: 908657. doi: 10.3389/fped.2022.908657
Mroczek Magdalena , Maniscalco Ignazio , Sendel Manon , Baron Ralf , Seifritz Erich , Nowak Albina Neuropsychiatric Symptoms and Their Association With Sex, Age, and Enzyme Replacement Therapy in Fabry Disease: A Systematic ReviewFrontiers in Psychiatry, v.13, 2022, DOI=10.3389/fpsyt.2022.829128
Ramaswami U, Parini R, Kampmann C, Beck M. Safety of agalsidase alfa in patients with Fabry disease under 7 years. Acta Paediatr. 2011 Apr;100(4):605-11. doi: 10.1111/j.1651-2227.2010.02101.x
Hopkin RJ, Jefferies JL, Laney DA, Lawson VH, Mauer M, Taylor MR, Wilcox WR; Fabry Pediatric Expert Panel. The management and treatment of children with Fabry disease: A United States-based perspective. Mol Genet Metab. 2016 Feb;117(2):104-13. doi: 10.1016/j.ymgme.2015.10.007
Mackels L, Servais L. The Importance of Early Treatment of Inherited Neuromuscular Conditions. J Neuromuscul Dis. 2024;11(2):253-274. doi: 10.3233/JND-230189. PMID: 38306060; PMCID: PMC10977423.
Madsen CV, Christensen EI, Nielsen R, Mogensen H, Rasmussen ÅK, Feldt-Rasmussen U. Enzyme Replacement Therapy During Pregnancy in Fabry Patients : Review of Published Cases of Live Births and a New Case of a Severely Affected Female with Fabry Disease and Pre-eclampsia Complicating Pregnancy. JIMD Rep. 2019;44:93-101. doi: 10.1007/8904_2018_129. Epub 2018 Aug 17. PMID: 30117110; PMCID: PMC6323029
Fernández P, Fernández SO, Gonzalez JGM, Fernández T, Fernández CC, Fernández SP. Enzyme Replacement Therapy in Pregnant Women with Fabry Disease: A Case Series. JIMD Rep. 2019;45:77-81. doi: 10.1007/8904_2018_141. Epub 2018 Nov 8. PMID: 30406505; PMCID: PMC6336548
Stepien KM, Broomfield A, Cole D, Deegan PB, Forshaw-Hulme S, Hughes D, Jovanovic A, Morris L, Muir A, Ramaswami U. Management of pain in Fabry disease in the UK clinical setting: consensus findings from an expert Delphi panel. Orphanet J Rare Dis. 2023 Jul 21;18(1):203. doi: 10.1186/s13023-023-02796-1. PMID: 37480023; PMCID: PMC10362568,