Эпилепсия и эпилептический статус у взрослых и детей
Список сокращений
АТФ – Аденозинтрифосфат
ГАМК – гамма-аминомасляная кислота
ДАЭ – детская абсансная эпилепсия
ИГЭ – идиопатическая генерализованная эпилепсия
КД – кетогенная диета
МВЭ – медиальная височная эпилепсия
МПЭЛ (ILAE) – Международная противоэпилептическая лига.
ФКД – фокальная кортикальная дисплазия
ПЭП – противоэпилептические препараты
ЭС – эпилептический статус
ЭССП – эпилептический статус судорожных приступов
ЦСЖ – цереброспинальная жидкость
цАМФ – циклический аденозинмонофосфат
ЦНС – центральная нервная система
ЧМТ – черепно-мозговая травма
ЭЭГ – электроэнцефалография
ЮАЭ – юношеская абсансная эпилепсия
ЮМЭ – юношеская миоклоническая эпилепсия
СИОЗС – селективные ингибиторы обратного захвата серотонина
АМРА – тип ионотропного рецептора глутамата (от англ. Α-Amino-3-Hydroxy-5-Methyl-4-Isoxazolepropionic Acid – название специфического агониста рецептора)
BDNF – нейротрофический фактор мозга (от англ. Brain-Derived Neurotrophic Factor)
CACNA1A – ген, кодирующий альфа-1а субъединицу потенциалзависимого кальциевого ионотропного канала P/Q типа (от англ. Calcium Channel, Voltage-Dependent, P/Q Type, Alpha-1a Subunit)
CHRNA4 – ген, кодирующий альфа-4 субъединицу холинергического никотинового ионотропного рецептора (от англ. Cholinergic Receptor, Neuronal Nicotinic, Alpha Polypeptide 4)
CHRNB2 – ген, кодирующий бета-2 субъединицу холинергического никотинового ионотропного рецептора (от англ. Cholinergic Receptor, Neuronal Nicotinic, Beta Polypeptide 2)
CLCN2 – ген, кодирующий хлорный потенциалзависимый ионный канал типа 2 (от англ. ChlorideChannel 2)
CRMP-5 – collapsingresponsemediatorprotein 5, белок-медиатор коллапсинового ответа 5
ENFL1, ENFL3 - эпилепсия с ночными лобными приступами типа 1 и типа 3 (от англ. Epilepsy, Nocturnal Frontal Lobe, 1; 3)
GABAA – тип А ионотропного рецептора гамма-аминомасляной кислоты (от англ. Gamma-Aminobutyric Acid)
GABAB - тип В ионотропного рецептора гамма-аминомасляной кислоты (от англ. Gamma-Aminobutyric Acid)
GABAaR – gamma-aminobutyricacidtypeAreceptor, рецепторы гамма-аминомасляной кислоты типа А
GABRA1 – ген, кодирующий альфа-1 субъединицу ионотропного рецептора ГАМК типа А (от англ. Gamma-Aminobutyric Acid Receptor, Alpha-1)
GABRG2 – ген, кодирующий гамма-2 субъединицу ионотропного рецептора ГАМК типа А (от англ. Gamma-Aminobutyric Acid Receptor, Gamma-2)
GAD65 – glutamicaciddecarboxylase, декарбоксилаза глутаминовой кислоты 65
GEFS+ – генерализованная эпилепсия с фебрильными судорогами Плюс (от англ. GeneralizedEpilepsyWithFebrileSeizuresPlus)
LEATs – long-termepilepsyassociatedtumors, опухоли, ассоциированные с длительно присутствующей эпилепсией
LGI1 - leucine-richglioma-inactivatedprotein 1, богатый лейцином инактивируемый глиомой протеин 1
KCNA1 – ген, кодирующий потенциалзависимый калиевый ионотропный канал, подсемейство А, тип 1 (от англ. Potassium Channel, Voltage-Gated, Shaker-Related Subfamily, Member 1)
KCND2 – ген, кодирующий калиевый потенциалзависимый ионотропный канал, подсемейство D, тип 2 (от англ. Potassium Voltage-Gated Channel, Shal-Related Subfamily, Member 2; KCND2
KCNMA1 – ген, кодирующий альфа-1 субъединицу калиевого кальций-активируемого ионного канала, подсемейства М (от англ. Potassium Channel, Calcium-Activated, Large Conductance, Subfamily M, Alpha Member 1)
KCNQ2, KCNQ3 – гены, кодирующие калиевые потенциалзависимые ионные каналы, подсемейство Q, типы 2 и 3 (от англ. Potassium Channel, Voltage-Gated, KQT-Like Subfamily, Member 2)
NMDA - тип ионотропного рецептора глутамата (от англ. N-Methyl-D-Aspartic Acid – название специфического агониста рецептора)
NDDI-E - Неврологический опросник депрессивного расстройства при эпилепсии
Se – чувствительность
Sp – специфичность
SCN1A – ген, кодирующий альфа-1 субъединицу натриевого потенциал-зависимого ионного канала (Sodium Voltage-Gated Channel, Alpha Subunit 1, сокращенное название от англ. Sodium Channel, Neuronal Type 1, Alpha Subunit)
SCN2A – ген, кодирующий альфа-2 субъединицу натриевого потенциал-зависимого ионного канала (Sodium Voltage-Gated Channel, Alpha Subunit 2, сокращенное название гена от англ. Sodium Channel, Neuronal Type 2, Alpha Subunit)
SCN1B – ген, кодирующий бета-1 субъединицу натриевого потенциалзависимого ионного канала (Sodium Voltage-Gated Channel, Beta Subunit 1, сокращение гена от англ. SodiumChannel, NeuronalType1, BetaSubunit)
Термины и определения
Абсанс – это разновидность генерализованного эпилептического приступа: немоторная форма. Характеризуется внезапной кратковременной (секунды-десятки секунд) утратой сознания, блокадой моторной активности и амнезией. Имеет специфический паттерн ЭЭГ. См. абсанс типичный и атипичный.
Абсанс атипичный – это абсанс с изменениями в тонусе, которые являются более выраженными, чем при типичном абсансе; начало и/или прекращение не являются внезапными, часто ассоциирован с медленной нерегулярной генерализованной пик-волновой активностью в ЭЭГ [1]. Абсанс атипичный – разновидность абсанса, простого или сложного, выделенного на основании электроэнцефалографических показателей: спайк-волновые разряды менее 3 Гц [2].
Абсанс простой – это абсанс с полной блокадой моторной активности, за редким исключением эпилептического миоклонуса век.
Абсанс сложный – это абсанс, во время которого выключение сознания сопровождается другими симптомами, выходящими на первый план: автоматизмами, обычно элементарными (абсанс автоматизмов), миоклониями (миоклонический абсанс), падением (атонический абсанс), упусканием мочи (автономно-вегетативный абсанс).
Абсанс субклинический (синоним фантомный) – это абсанс, длящийся слишком короткое время (2 - 4 с), чтобы можно было заметить выключение сознания, регистрируется только электрографически.
Абсанс типичный – разновидность абсанса простого или сложного, сопровождающегося в ЭЭГ двусторонними синхронными симметричными разрядами комплексов пик-волна частотой 3 Гц. Легко провоцируется гипервентиляцией. Клинически характеризуется внезапным началом, прерыванием текущей активности, отсутствующим взглядом, возможна кратковременная девиация глаз. Обычно пациент не реагирует на обращение к нему. Продолжительность – от нескольких до 30 секунд с очень быстрым восстановлением. Абсанс по определению является приступом с генерализованным дебютом. Термин не является синонимом «отсутствующего взгляда», который также может встречаться при судорожных приступах.
Автоматизм - более или менее скоординированная двигательная активность, которая обычно возникает на фоне расстройства когнитивных функций, чаще с последующей амнезией. Часто напоминает контролируемые движения и может представлять собой измененную двигательную активность, имевшую место до приступа.
Автоматизм эпилептический – непроизвольная двигательная активность, более или менее координированная и адаптированная. Обычно проявление фокального приступа. Приступы фокального автоматизма имеют следующие категории:
Орофациальный: чмокание губами, поджатие губ, жевание, глотание, цоканье, моргание глаз.
Мануальный: односторонние или двусторонние ощупывания, постукивание, манипуляционные или исследовательские движения руками.
Стопный: двусторонние или односторонние движения ступней/ног, которые могут включать расхаживания, ходьбу или бег. Движение больше напоминает нормальные движения по амплитуде и менее хаотичные и быстрые по сравнению с движениями, наблюдаемыми при фокальных гиперкинетических приступах с вовлечением ног.
Персеверативный: движение состоит из несоответствующего повторения тех движений, которые были у человека до приступа.
Вокальный: одиночные или повторяющиеся звуки, такие как визг или хрюканье.
Вербальный: отдельные или повторяющиеся слова, фразы или короткие предложения.
Сексуальный: сексуальное поведение.
Неклассифицированный (другой): автоматизм может включать кивание головой, раздевание и ряд других автоматических движений
Диапазон проявлений крайне широк: от элементарных автоматизмов до автоматизмов действий и деятельности (поведение автоматическое эпилептическое – в настоящее время термин не рекомендован). Развиваются по механизму высвобождения или прямой активации эпилептической активности соответствующих моторных структур. Если автоматизм есть проявление, ограниченное моторикой самого больного, он квалифицируется как аутомоторный приступ (автоматизмы алиментарные, автоматизмы мимические, автоматизмы жестов, автоматизмы речи). Часто напоминают произвольные движения и могут существовать как неадекватное продолжение преиктальной моторной активности. Постприступные двигательные расстройства являются скорее проявлением неосознанной нецелевой агрессии и могут носить разрушительный характер.
Амавроз эпилептический – преходящий амавроз, чаще являющийся проявлением приступа затылочной эпилепсии. Может быть как семиотикой самого приступа, так и симптомом постиктального выпадения.
Атонический приступ - внезапная потеря или снижение мышечного тонуса без видимого предшествующего миоклонического или тонического компонента длительностью ~ 1 - 2 с, включая мышцы головы, туловища, лица или конечностей.
Аура - внезапный субъективный феномен, специфичный для конкретного пациента, который может предшествовать приступу. В классификации 2017 года рекомендовано использование термина: Фокальный без нарушения сознания.
Билатеральные тонико-клонические приступы с фокальным началом - тип приступов с фокальным дебютом, с сохранением или нарушением сознания, могут быть моторными или немоторными, с последующей развивающейся билатеральной тонико-клонической активностью. Предыдущий термин – «вторично-генерализованные приступы с парциальным началом».
Билатеральный приступ - вовлекающий левую и правую стороны, хотя проявления билатеральных приступов могут быть как симметричными, так и асимметричными.
Вегетативные (автономные) приступы - явное нарушение функции вегетативной нервной системы, включающее изменение диаметра зрачков, потоотделение, изменение тонуса сосудов, терморегуляции, расстройства функции ЖКТ и сердечно-сосудистой системы.
Версивный приступ - длительное насильственное сопряженное вращение глаз, головы и туловища или их отклонение латерально от центральной оси.
Галлюцинации - составление композиции восприятия без соответствующих внешних стимулов, включая зрительные, слуховые, соматосенсорные, обонятельные и вкусовые стимулы.
Геластический приступ – внезапные эпизоды смеха или хихиканья, обычно без соответствующего аффективного фона.
Геластическая эпилепсия, синоним - гелолепсия – эпилепсия смеха.
Генерализованный приступ - приступ, развивающийся в какой-то момент в нейрональной сети одного полушария, но с последующим быстрым вовлечением билатерально распространенных нейрональных сетей.
Генерализованный тонико-клонический приступ - билатеральные симметричные, иногда асимметричные тонические сокращения и затем двустороннее клоническое подергивание мышц, обычно сочетающееся с вегетативными феноменами и нарушением сознания. Эти приступы с самого начала охватывают нейрональные сетевые структуры обоих полушарий.
Гипсаритмия – характерный межприступный ЭЭГ-паттерн, который обычно, но не всегда, наблюдается у грудных детей с синдромом инфантильных спазмов (синдромом Веста). Представляет собой диффузную продолженную нерегулярную медленноволновую активность очень высокой амплитуды (более 300 мкВ) в сочетании с мультирегиональными спайками и острыми волнами, обычно высокой степени дезорганизованности и асинхронности. Наиболее выражен в фазу медленного сна и отсутствует или минимально выражен во время REM сна. Варианты включают асимметрию, преобладание фокального фокуса (на фоне диффузных изменений), эпизоды подавления биоэлектрической активности или ее фрагментации, усиленную периодичность и сохранение межполушарной синхронизации (все варианты относятся к термину «модифицированная гипсаритмия»).
Дакристический приступ - приступ плача, не обязательно связанный с эмоциональными проявлениями.
Джексоновский приступ - традиционный термин, обозначающий распространение клонических подергиваний унилатерально через смежные части тела.
Дистонический приступ – приступ с устойчивыми сокращениями как синергических, так и антагонистических мышц, вызывающих атетоидные или скручивающие движения, которые могут формировать неестественные позы.
Иктальный – от английского ictus – приступ: связанный с приступом.
Иктальный ЭЭГ-паттерн (синоним: ЭЭГ-паттерн эпилептического приступа) - феномен, состоящий из повторяющихся эпилептиформных разрядов с частотой > 2 в секунду и/или характерного паттерна с квазиритмичной пространственно-временной эволюцией (например, постепенным изменением частоты, амплитуды, морфологии и локализации), продолжающегося как минимум несколько секунд (обычно >10сек). Кроме того, к иктальным паттернам относят электродекремент и низковольтажную быструю активность, которые могут длиться менее 10 с. Частые интериктальные эпилептиформные разряды обычно не связаны с клиническими приступами и их следует отличать от электрографического паттерна приступа. Комментарий: ЭЭГ-паттерны приступа, которые не сопровождаются клиническими эпилептическими проявлениями, называют электрографическими субклиническими приступами.
Интериктальный – межприступный (см. иктальный).
Клонический приступ – приступ симметричного или асимметричного подергивания, которое регулярно повторяется и включает одни и те же группы мышц.
Когнитивный приступ - относится к мышлению и высшим мозговым функциям, таким как язык, пространственное восприятие, память и праксис. В соответствии с глоссарием 2001 года, имел значение «психический».
Марш джексоновский – характер распространения клоний или парестезий по смежным отделам тела при джексоновском, то есть соматомоторном или соматосенсорном приступе, по новой терминологии – фокальном моторном или немоторном приступе.
Миоклонико-атонический - генерализованный тип приступов с миоклоническими подергиваниями, предшествующими атоническому моторному компоненту. Этот тип приступов ранее назывался миоклонико-астатическим.
Миоклонико-тонико-клонический - одно или несколько билатеральных подергиваний мышц туловища, с последующим развитием тонико-клонического приступа. Первоначальные подергивания можно рассматривать как короткий период клонуса или миоклонуса. Приступы такого типа характерны для юношеской (синоним ювенильной) миоклонической эпилепсии.
Миоклонический приступ - внезапное, краткое (< 100 мс) непроизвольное одиночное или множественное сокращение мышц или групп мышц с переменной топографией (аксиальная, проксимальная, мышцы туловища, дистальная). При миоклонусе движения повторяются менее регулярно и с меньшей продолжительностью, чем при клонусе.
Миоклония век - подергивание век с частотой не менее 3 раз в секунду, как правило, с девиацией глаз вверх, длящееся < 10 с, часто провоцируется закрытием глаз. В части случаев может сопровождаться кратковременной потерей ориентации.
Моторный - любая форма вовлечения мускулатуры. Моторная активность может заключаться как в повышенном сокращении (положительная), так и в сниженном сокращении мышц (отрицательная) при продуцировании движений.
Нарушение сознания («impairment of consciousness») - смотри «нарушенное сознание».
Нарушение осознанности («impaired awareness») - смотри «сознание (awareness)». Ослабленное или утраченное сознание является признаком фокального приступа с нарушением осознанности, ранее называвшегося сложным парциальным приступом.
Неклассифицированный приступ - может применяться по отношению к типу приступов, который не описан в Классификации ILAE 2017 г. по причине недостаточной информации либо необычных клинических проявлений. Если приступ не классифицирован в связи с недостаточной информацией о его начале, он может быть ограниченно классифицирован на основании доступных для интерпретации данных.
Немоторный приступ - фокальный или генерализованный приступ, при которых не проявляется двигательная (моторная) активность.
Неподвижность - смотри «заторможенность поведенческих реакций».
Нистагм эпилептический – окулоклонический эпилептический приступ.
Обморок судорожный (синоним: судорожный синкопе)– классический пример вторичного вовлечения мозга в приступ вследствие экстрацеребральной патологии, чаще всего кардиогенной. Судорожный компонент является проявлением глубокого обморока. Он требует дифференциального диагноза с эпилептическим приступом. При этом данные осмотра терапевтом и неврологом, включая холтер-ЭКГ и ЭЭГ-мониторирование, могут оказаться недостаточными. В этих случаях высоко значимой является длительная пассивная ортостатическая проба, при недостаточной информативности - тилт-тест. Показана возможность коморбидности по эпилепсии и обморокам кардиоингибиторного и вазодепрессорного типа.
Остановка поведенческой деятельности - заторможенность или пауза в деятельности, застывание, неподвижность, свойственная приступам с заторможенностью поведенческих реакций.
Отсутствие ответа - неспособность адекватно отреагировать движением или речью на предъявляемый стимул.
Очаг эпилептический – популяция эпилептических нейронов, продуцирующих гиперсинхронный разряд и являющихся источником эпилептического приступа. В межприступном периоде может быть обнаружен в ЭЭГ в виде фокальных эпилептических электрографических феноменов – спайка, полиспайка, медленного спайка (острой волны) и/или указанных феноменов с последующей медленной волной.
Очаг эпилептогенный – участок органического повреждения мозга, в котором сохранившиеся нейроны не удерживают поляризацию (эпилептический нейрон), вследствие чего создается эпилептический очаги развитие эпилепсии.
Паралич Тодда – синоним термина послеприступный эпилептический паралич.
Пароксизм электроэнцефалографический эпилептический – волна или группа волн, отражающие на ЭЭГ эпилептический разряд, которые возникают и исчезают внезапно и четко отличаются по частоте, морфологии и амплитуде от фоновой активности в ЭЭГ, характеризуясь в целом патогномоничными эпилептическими паттернами – спайк, медленный спайк (острая волна), комплекс спайк-волна. Чаще используется синоним электрографический эпилептический разряд. Согласно ILAE - феномен ЭЭГ, который внезапно начинается, быстро достигает максимума и внезапно прекращается. Четко выделяется из фоновой активности. Комментарий: обычно используют при описании эпилептиформных паттернов и паттернов приступа.
Предрасположенность к эпилепсии – конституциональное или приобретенное состояние, предрасполагающее субъекта к различным типам эпилептических приступов. В настоящее время устойчивое предрасположение является ключевым словом в дефиниции эпилепсии. Предполагается облигатное участие этого фактора даже для реализации приступов после органического поражения мозга. Показана гетерогенность генетической предрасположенности, которая может иметь избирательный характер, например, предрасположенность к судорожным приступам (предрасположение судорожное).
Приступ (припадок) – событие церебрального происхождения, наступающий у человека внешне на фоне видимого здоровья или при внезапном ухудшении хронического патологического состояния. Проявляется внезапно возникающими преходящими патологическими феноменами – двигательными, сенсорными, вегетативными или психическими в результате временной дисфункции всего мозга или его отдела. Наиболее практично квалифицировать эти приступы по их механизму:
а) приступы эпилептической природы, вызываемые чрезмерными разрядами нейронных популяций;
б) церебральные приступы аноксического и анокси-ишемического генеза;
в) церебральные приступы токсического происхождения (столбняк, стрихнин и др.);
г) церебральные приступы метаболического происхождения (гипогликемические, гипокальциемические и др.);
д) церебральные приступы аутоиммунной природы;
е) церебральные приступы гипнической природы (нарколептические, каталептические);
ж) психогенные неэпилептические приступы [1, 3], или – на языке психиатров – конверсионные приступы (синоним - функциональные неэпилептические приступы).
з) церебральные приступы неопределенного или сложного происхождения, при которых действующими являются несколько факторов (Примечание: каталептические и нарколептические приступы являются исключением, их принадлежность к гипнотическим есть результат их связи со сном и, возможно, с участием в реализации некоторых общих механизмов. Краткое определение приступа см. приступ (Глоссарий МПЭЛ, 2017).
Приступ (Глоссарий МПЭЛ, 2017) - преходящее появление признаков и/или симптомов, связанных с аномальной избыточной или синхронной активностью нейронов в головном мозге.
Приступ автоматизма эпилептический – приступ непроизвольной более или менее координированной автоматизированной деятельности, проявление генерализованного (сложный абсанс – абсанс автоматизмов: автоматизмы элементарны, осознание приступа отсутствует) или сложного фокального эпилептического приступа: автоматизмы более сложны, проходят на фоне помраченного сознания.
Приступ аутоиндуцированный – редкая разновидность эпилептического приступа, произвольно вызываемого самим пациентом.
Приступ аффективный эпилептический – эпилептический приступ, начальные проявления которого выражаются в аффекте: немотивированном страхе или, наоборот, в радости, удовольствии или даже экстазе.
Приступ большой эпилептический, приступ grand mal (большой судорожный приступ). В условиях первичного амбулаторного приема как врачи, так и пациенты до настоящего времени пользуются этим термином для дальнейшей ориентации разграничения генерализованного судорожного приступа от других приступов. (см. Глоссарий МПЭЛ, 2017).
Приступ в виде «цифры 4» - приступы, характеризующиеся разгибанием одной руки перпендикулярно туловищу (обычно контралатеральной эпилептогенной зоне в головном мозге) и сгибанием в локте другой руки, образующими цифру «4».
Приступ версивный эпилептический – приступ поворота (версии): 1) глазных яблок клонического характера – окулоклонический эпилептический приступ, эпилептический нистагм; 2) то же тонического характера – глазодвигательный эпилептический приступ; 3) глаз, головы и туловища – адверсивный эпилептический приступ; 4) то же с последующим вращением туловища вокруг своей оси – вращательный эпилептический приступ.
Приступ генерализованный эпилептический – эпилептический приступ, характеризующийся 1) клинически: нарушением сознания, массивными автономными (вегетативными) проявлениями, сопровождающимися или не сопровождающимися моторными феноменами, в частности, судорожными, вовлекающими обе стороны тела одновременно. В зависимости от наличия или отсутствия судорог различают генерализованные судорожные эпилептические приступы (тонико-клонические, клонико-тонико-клонические, тонические, двусторонние массивные эпилептические миоклонии) и бессудорожные (абсансы простые и сложные) атонические приступы; 2) электроэнцефалографически: двусторонними синхронными и симметричными разрядами эпилептического приступа. Эти приступы, особенно судорожные, могут начинаться с фокальных проявлений, в этих случаях используется термин билатеральные тонико-клонические. При развитии генерализованного приступа из фокального - до 2017 года использовался термин вторично генерализованного эпилептического приступа.
Приступ децеребрации – неэпилептический тонический приступ – опистотонус, связанный с высвобождением тоногенных структур мозгового ствола, обычно в результате острой или декомпенсации текущей внутримозговой гипертензии.
Приступ дисмнестический – разновидность фокального эпилептического приступа, наиболее значимым проявлением которого являются нарушения памяти, как правило, в результате нейронных разрядов с участием старой коры (гиппокамп). Сюда входят более мягкие нарушения – ощущение уже виденного, уже слышанного, уже пережитого, никогда не виденного, никогда не слышанного, никогда не пережитого – и более глубокие: галлюцинаторные эпилептические приступы, панорамное видение.
Приступ идеаторный эпилептический – разновидность фокального эпилептического приступа, основным или единственным признаком которого служит навязчивая идея (насильственное мышление эпилептическое). Идея может оставаться той же, которая была при наступлении приступа, или новой, появившейся уже в ходе приступа (паразитическая идея).
Приступ ипсилатеральный эпилептический – разновидность версивного эпилептического приступа, характеризующегося поворотом в сторону полушария, в котором находится эпилептический очаг.
Приступ катамениальный эпилептический – приступ, возникающий в определенную фазу менструального цикла, в прежнем понимании – приступ в дни менструации или следующие после нее дни. Синоним – менструальный эпилептический приступ.
Приступ малый эпилептический, приступ petit mal – бессудорожный генерализованный эпилептический приступ. Также термин, используемый до сих пор на первичном амбулаторном приеме врачами и пациентами для ориентировочного разграничения любого другого приступа с большим судорожным приступом.
Приступ окулоклонический - см. версивный эпилептический приступ.
Приступ перемежающийся эпилептический – альтернирующие гемиконвульсии, принадлежность новорожденных и младенцев.
Приступ произвольно вызванный эпилептический – синоним термина приступ автоиндуцируемый эпилептический.
Приступ прокурсивный (лат procursum – выбегать вперед) эпилептический – вариант фокального эпилептического приступа, характеризующегося пропульсивными автоматизмами (ходьба, бег вперед, несмотря на препятствия) на фоне измененного сознания.
Приступ рефлекторный эпилептический – разновидность эпилептического приступа, вызываемого всегда определенными стимулами: 1) испугом, 2) элементарными сенсорными стимулами и 3) мышлением. См. эпилепсия рефлекторная.
Приступ с «позой фехтовальщика» - тип фокального моторного приступа с вытягиванием одной руки и сгибанием в локте другой, имитирующий фехтование. Также называется «приступом дополнительной моторной зоны».
Приступ случайный (синоним ситуационно-обусловленный) эпилептический: 1) эпилептический приступ, возникающий у здорового человека под влиянием случайных причин: длительной депривации сна, интоксикации инсектицидами, медикаментами (бемегрид и др.) и т.д. и 2) приступы, проявляющиеся только в острой стадии органического поражения мозга – нарушения мозгового кровообращения, черепно-мозговой травмы и т.д.: острый симптоматический эпилептический приступ.
Приступ фебрильный – приступ, возникающий при подъеме температуры.
Приступ эпилептический – приступ, возникающий в результате чрезмерных нейронных разрядов. В соответствии с их клинико-электроэнцефалографической характеристикой различают генерализованные эпилептические приступы, фокальные эпилептические приступы, односторонние эпилептические приступы и билатеральные тонико-клонические приступы с фокальным началом, а также неклассифицированные эпилептические приступы.
Продром (предвестник) эпилептический. Симптомы, которые появляются за значимое время (десятки минут, несколько часов или дней до появления приступа у пациентов с эпилепсией (изменение настроения, головная боль, астения и др.)).
Психоз эпилептический – термин, обозначающий психотические проявления у пациентов с эпилепсией. Хронический эпилептический психоз – галлюцинаторно-параноидный по характеру, проявляется преимущественно в виде бредовых идей, обычно мистического и религиозного содержания. Острый эпилептический психоз обычно выступает вне зависимости от приступов в виде делириозных атак или шизофренических эпизодов, длящихся дни и недели. Как правило, эпилептические психозы развиваются у больных с височной эпилепсией. Альтернативный психоз, или насильственная нормализация электроэнцефалограммы: фактическое замещение приступов психозом с одновременным исчезновением эпилептиформной активности в ЭЭГ. Это случается на фоне спонтанной или ятрогенной ремиссии приступов.
Разрешение эпилепсии - эпилепсия считается разрешившейся у достигших определенного возраста пациентов с возрастзависимыми эпилептическими синдромами, либо при отсутствии эпилептических приступов в течение 10 лет у пациентов, в том числе не использовавших противоэпилептические препараты (ПЭП) за последние 5 лет.
Разряд - колебания кривой с не более чем тремя фазами (пересекают изолинию не более двух раз) или длительностью 0,5 с или меньше, независимо от количества фаз. Термин, интерпретирующий колебания потенциалов действия и постсинаптических потенциалов, обычно используют для обозначения межприступных паттернов и паттернов приступа (см. Эпилептиформный паттерн, ЭЭГ-паттерн эпилептического приступа).
Разряд генерализованный эпилептический – электроэнцефалографический пароксизм, имеющий в скальповой ЭЭГ билатеральный синхронный и симметричный характер. Разряд иктальный эпилептический – синоним термина разряд приступа (электроэнцефалографический).
Разряд межприступный эпилептический (электроэнцефалографический) (синоним эпилептиформный паттерн – электроэнцефалографический эпилептический разряд, регистрируемый у больных эпилепсией в промежутках между приступами. Описывает компоненты, которые выделяются из фоновой активности, с характерной спайковой морфологией, как правило (но не всегда) регистрируемые в интериктальной ЭЭГ больных эпилепсией. Эпилептиформные паттерны должны удовлетворять как минимум 4 из 6 критериев: 1) ди- или трифазная острая волна или спайк, 2) отличие периода волны от фоновой активности (меньше или больше периода волн в фоне), 3) асимметрия формы волны: круто нарастающая восходящая фаза и менее крутая нисходящая, 4) за острой волной следует связанная с ней медленная волна, 5) эпилептиформный разряд нарушает структуру фоновой активности, 6) расположение негативной и позитивной фаз потенциала предполагает наличие источника в веществе головного мозга, соответствующего радиальному, косому или тангенциальному диполю. Персистирующая эпилептиформная активность дезорганизует работу тех регионов мозга, где она регистрируется, в результате чего, особенно у детей, иногда развиваются тяжелые расстройства: когнитивные, мнестические, речевые, а также девиантное и даже асоциальное поведение – эпилептическая энцефалопатия в современном клинико-электроэнцефалографическом значении этого термина.
Разряд субклинический (электроэнцефалографический) эпилептический – разряд эпилептиформной активности, не сопровождающийся клиническими проявлениями. Разряд эпилептический – нейронный разряд в результате одновременной синхронной и синфазной, а также часто самоподдерживающейся активации большой популяции нейронов. Эти разряды не всегда можно записать со скальпа. Шансы их регистрации возрастают с увеличением непрерывной длительной записи ЭЭГ (суточный, двух-, трехсуточный ЭЭГ-мониторинг).
Распространение - распространение судорожной активности из одного мозгового центра на другой или вовлечение дополнительных сетевых структур мозга.
Реакция - способность адекватно отреагировать движением или речью на предъявленный стимул.
Ритм вовлечения эпилептический – ЭЭГ-ритм, вначале низкоамплитудный с частотой около 14 в с, постепенно замедляющийся и возрастающий по амплитуде. Этот ритм, который распространяется по всему скальпу, является электрографическим коррелятом тонической фазы тонико-клонических судорожных приступов, самих тонических приступов и некоторых атипичных абсансов. Синоним: рекрутирующий ритм.
Сенсорный приступ - субъективно воспринимаемое ощущение, не вызванное соответствующими стимулами во внешнем мире.
Синдром гемиконвульсии–гемиплегии-эпилепсии – является редким последствием фокального моторного статуса младенчества и раннего детства. Начальным проявлением является фокальный клонический эпилептический статус в контексте фебрильного заболевания у ребенка 4 лет и младше. За острой фазой следует гемиатрофия полушария с последующим развитием фармакорезистентных фокальных приступов и гемипареза.
Сознание в смысле «разумение» - как субъективные, так и объективные проявления состояния рассудка, включающие осознание себя как уникальной сущности, восприятие, реакцию и память.
Сознание в смысле бодрствование - осознание себя или способность ориентироваться в окружающем пространстве.
Спазм - смотри «эпилептический спазм».
Спазм инфантильный – очень кратковременный эпилептический приступ, наблюдающийся у детей первых двух лет жизни, страдающих энцефалопатией развития и эпилептической, сочетающийся с задержкой развития и характерным межприступным ЭЭГ-паттерном – гипсаритмией. См. синдром инфантильных спазмов, спазм эпилептический.
Статус эпилептический – состояние пролонгированного приступа или повторяющихся приступов, в интервалах между которыми состояние больного не возвращается к исходному. Это результат отказа механизмов, ответственных за прекращение, либо инициация механизмов, ведущих к аномально пролонгированным приступам после временной точки t1 (время начала лечения), которые могут иметь долгосрочные последствия после рубежа t2 (время начала долгосрочных изменений), включающие нейрональную смерть, нейрональное повреждение, перестройку нейронных связей. Временные параметры: генерализованный тонико-клонический эпилептический статус t1 – 5 мин, t2 – 30 мин, фокальный эпилептический статус t1 – 10 мин, t2 – более 60 мин, статус абсансов t1 – 10 - 15 мин, t2 – неизвестно.
Тонико-клонический приступ - последовательность, состоящая из фазы тонического сокращения, за которой следует клоническая фаза.
Тонический приступ - устойчивое нарастающее сокращение мышц продолжительностью от нескольких секунд до нескольких минут.
Фокальный приступ - возникающий в нейросетевых структурах, ограниченных одним полушарием. Он может быть локализован или иметь более широкое распространение. Фокальные приступы могут возникать в подкорковых структурах.
Эклампсия – наиболее тяжелое позднее осложнение беременности, может развиваться в родах и послеродовом периоде. Основная клиническая триада: отеки, альбуминурия и гипертония. К ним присоединяются осложнения со стороны ЦНС – генерализованные судорожные приступы и кома.
Эмоциональный приступ - приступы с эмоциями или появлением эмоций как ранней характерной черты, таких как страх, внезапная (необоснованная) радость или эйфория, смех (геластический) или плач (дакристический).
Эпилепсия – хроническое заболевание головного мозга различной этиологии, характеризующееся повторными приступами, возникающими в результате чрезмерных нейронных разрядов (эпилептические приступы) и сопровождающееся разнообразными клиническим и параклиническими симптомами (Гасто А., 1975). Эпилепсия – заболевание мозга, характеризующееся стойким предрасположением к генерированию эпилептических приступов и сопровождающееся нейробиологическими, когнитивными, психологическими и социальными последствиями этого состояния (МПЭЛ 2005 - Berg A.T. et al., 2010). Согласно этому определению, для диагноза эпилепсии достаточно одного приступа. Эпилепсия – заболевание головного мозга, определяемая любым из следующих условий: 1) по крайней мере, два неспровоцированных (или рефлекторных) приступа, с интервалом > 24 ч; 2) один неспровоцированный (или рефлекторный) приступ и вероятность повторения приступов, близкая к общему риску рецидива (≥ 60%) после двух спонтанных приступов, в последующие 10 лет; 3) диагноз эпилептического синдрома (≥ 60% - следует трактовать как высокую вероятность рецидива) [3].
Эпилепсия генерализованная – форма эпилепсии, характеризующаяся исключительно (первично) генерализованными эпилептическими приступами с их электрографическими коррелятами.
Эпилепсия генетическая – результат известной или предполагаемой генетической мутации, при которой приступы являются стержневым симптомом заболевания. Молекулярная генетика позволяет идентифицировать причинные мутации большой группы эпилептических генов, часто возникающих de novo.
Эпилепсия идиопатическая – термин, применявшийся в течение длительного времени для обозначения эпилепсии с первично генерализованными приступами, наличием симметричных синхронных эпилептических паттернов в ЭЭГ и отсутствием значимых изменений психики и неврологического статуса. В настоящее время заменен термином эпилепсия генетическая для случаев, где генетическая этиология доказана или наиболее вероятна.
Эпилепсия иммунная – одно из проявлений иммунных заболеваний или аутоиммунно опосредованных воспалений в ЦНС. Количество таких энцефалитов быстро растет, особенно в связи с возрастающей доступностью тест-систем.
Эпилепсия инфекционная – эпилепсия, являющаяся прямым следствием конкретного инфекционного заболевания, и эпилептические приступы доминируют в их клинической картине. Наряду с некоторыми повсеместно распространенными инфекциями, например, респираторными, в мире существуют эндемичные районы по отдельным видам инфекций, в частности, по церебральной малярии, цистицеркозу, клещевой инфекции и др.
Эпилепсия метаболическая – разновидность симптоматической эпилепсии, возникшей в результате метаболических нарушений: витаминов и электролитов, например, недостаток пиридоксина (гипопиридоксинемия), кальция (гипокальциемия), биотина, фолиевой кислоты и цианокобаламина; органических кислот, в частности, D-глюконовая ацидемия, пропионовая ацидемия, дефицит сульфитоксидазы, дефицит фруктозо-1-6-дифосфатазы, а также аминоацидопатии (фенилкетонурия) и др.; углеводного, в частности, гипогликемия; жирового, например, липоидоз; водно-электролитного (эксикоз) и других (болезнь Менкеса, болезнь Краббе, митохондриальные болезни).
Эпилепсия миоклоническая – термин, относящийся к формам эпилепсии, где миоклонии являются единственным или преобладающим феноменом, таким образом, термин имеет собирательное значение и не является диагнозом. Термин охватывает широкий круг заболеваний от новорожденности (синдром Айкарди) до юности (юношеская миоклоническая эпилепсия) и более позднего возраста (некоторые миоклонус-эпилепсии) с разной клиникой и прогнозом.
Эпилепсия неизвестной этиологии – эпилепсия, причина которой пока не установлена. Это может быть связано с отсутствием полноценных диагностических средств, например, ЭЭГ, у больных с семиологией, такой же, как при лобной эпилепсии, или с другими причинами.
Эпилепсия рефлекторная – форма эпилепсии, при которой все или большая часть приступов провоцируются определенными стимулами. Синоним: эпилепсия стимулозависимая. См. приступ рефлекторный эпилептический.
Эпилепсия структурная – эпилепсия, при которой электроклинические анормальности вместе со структурными, объективируемыми посредством методов нейровизуализации, являются обоснованием заключения об обусловленности эпилептических приступов визуализируемыми изменениями. Структурная этиология может быть приобретенной – инсульт, травма, инфекция – или генетической, такой как мальформации кортикального развития.
Эпилепсия фокальная - это эпилепсия с одним или несколькими фокусами, а также эпилепсии с вовлечением одной гемисферы головного мозга. Для них характерен целый спектр клинических проявлений и фокальные эпилептиформные разряды на ЭЭГ.
Эпилептический спазм - приступ с внезапным сгибанием, разгибанием или чередованием сгибания и растяжения преимущественно проксимальных мышц и мышц туловища, которое обычно более длительное, чем миоклонус, но не такое длительное, как тонический приступ. Могут возникнуть гримасы, кивки головы или мелкие движения глаз. Эпилептические спазмы часто развиваются в виде кластеров. Инфантильные спазмы являются наиболее известной формой, однако спазмы могут возникать в любом возрасте.
Эпилептический – термин, обозначающий все специфические проявления, связанные с эпилептическим приступом – как клинические, так и электрографические (ЭЭГ).
Эпилептогенный – способный вызвать эпилептический приступ. Это могут быть: 1) органические изменения мозга, такие как фокальная корковая дисплазия, посттравматический очаг и пр. в зависимости от выраженности эпилептического предрасположения; 2) различные химические вещества, включая медикаменты, обладающие проконвульсивными свойствами: камфора, цефалоспорины и т.д.; 3) воздействие электрическим током: «электрошоковая терапия», последнее, кстати, даже в настоящее время применяется в отдельных случаях при сверхрезистентном эпилептическом статусе.
1. Краткая информация по заболеванию или состоянию (группы заболеваний или состояний)
1.1 Определение заболевания или состояния (группы заболеваний или состояний)
1.1. Определение заболевания или состояния (группы заболеваний или состояний)
1.1.1. Определение эпилепсии
Эпилепсия – заболевание головного мозга, определяемая любым из следующих условий: 1) по крайней мере, два неспровоцированных (или рефлекторных) приступа, с интервалом > 24 ч; 2) один неспровоцированный (или рефлекторный) приступ и вероятность повторения приступов, близкая к общему риску рецидива (≥ 60%) после двух спонтанных приступов в последующие 10 лет; 3) диагноз эпилептического синдрома,
(≥ 60% - следует трактовать как высокую вероятность рецидива) [3].
1.1.2. Определение эпилептического статуса
Эпилептический статус – состояние пролонгированного приступа или повторяющихся приступов, в интервалах между которыми состояние больного не возвращается к исходному. Это результат отказа механизмов, ответственных за прекращение, либо инициация механизмов, ведущих к аномально пролонгированным приступам после временной точки t1 (время начала лечения), которые могут иметь долгосрочные последствия после рубежа t2 (время начала долгосрочных изменений), включающие нейрональную смерть, нейрональное повреждение, перестройку нейронных связей. Временные параметры: тонико-клонический эпилептический статус t1 – 5 мин, t2 – 30 мин, фокальный эпилептический статус t1 – 10 мин, t2 – более 60 мин, статус абсансов t1 – 10 - 15 мин, t2 – неизвестно [4].
1.2 Этиология и патогенез заболевания или состояния (группы заболеваний или состояний)
1.2.Этиология и патогенез заболевания или состояния (группы заболеваний или состояний)
1.2.1. Этиология эпилепсии
Эпилепсия – полиэтиологичное заболевание. В соответствии с классификацией эпилепсий Международной противоэпилептической лиги 2017 года [5], все формы эпилепсии подразделяются по этиологии на 6 категорий:
· генетические;
· структурные;
· метаболические;
· инфекционные;
· иммунные;
· с неизвестной причиной.
Определение этиологии эпилепсии играет решающую роль в выборе тактики ведения и лечения пациента. В ряде случаев у пациента может быть сочетание нескольких этиологических факторов, например, структурного и генетического. В.А. Карлов рекомендует рассматривать этиологические факторы как факторы риска эпилепсии, которые могут быть реализованы только при наличии наследственного предрасположения [6].
Генетические эпилепсии
В генетическую группу включено большое количество заболеваний, хромосомных и генных, как моногенных, так и полигенных, при которых эпилепсия может быть единственным проявлением заболевания или она входит в структуру заболевания наряду с другими симптомокомплексами.
Наследственные эпилепсии - группа генетически гетерогенных заболеваний, возникающих в результате мутаций в генах, количественных или структурных перестройках хромосом. В зависимости от этиологии можно выделить три основные группы наследственных эпилепсий: моногенные заболевания и синдромы, хромосомные синдромы и мультифакторные эпилепсии. Существует несколько групп моногенных заболеваний, в структуре симптомокомплекса которых отмечаются судороги: изолированные моногенные эпилепсии, моногенные синдромы и пороки развития мозга, дегенеративные заболевания нервной системы, наследственные болезни обмена веществ. В настоящее время идентифицировано более 700 генов, мутации в которых приводят к возникновению моногенных судорог [21].
Изолированные моногенные эпилепсии включают группы: ранних эпилептических младенческих энцефалопатий, миоклонус-эпилепсий детского и юношеского возраста, генерализованных эпилепсий с фебрильными судорогами плюс, доброкачественных фебрильных судорог височных и лобных эпилепсий. Каждая из этих групп насчитывает несколько генетических вариантов, обусловленных мутациями в отдельных генах [22].
Возникновение эпилепсий наблюдается также у больных с количественными и структурными перестройками хромосом. Идентифицировано несколько сотен хромосомных синдромов, сопровождающихся судорогами.
Значительная доля заболеваний приходится также на мультифакторные эпилепсии, возникающие при совместном действии наследственных факторов и факторов внешней среды. Наследственные факторы, в виде полиморфизмов в нескольких генах, формируют предрасположенность к возникновению судорог, которая реализуется под действием факторов внешней среды (травмы, инфекции, стресс и др.).
Установление этиологического фактора наследственного заболевания или синдрома, в большинстве случаев, является сложной задачей, так как требует использования различных биохимических и молекулярно-генетических методов исследования. Однако, обнаружение гена или хромосомной перестройки, ответственных за их возникновение, необходимо не только для уточнения диагноза, определения характера течения заболевания и эффективности его терапевтической и хирургической коррекции, но и для расчета риска рождения больного ребенка в отягощенной семье и планирования профилактических мероприятий [23].
Особенности клинических проявлений идиопатических и синдромальных вариантов моногенных эпилепсий.
Предположить наличие моногенного варианта идиопатических эпилепсий возможно в следующих случаях:
- наличие нескольких членов семьи, страдающих эпилепсией;
- отсутствие провоцирующего фактора возникновения судорог (инфекции, травмы и др);
- фармакорезистентность судорог;
- отсутствие значимой очаговой неврологической симптоматики у пациентов с судорогами.
В большинстве случаев у пациентов с моногенными вариантами эпилепсий судороги возникают после периода нормального психомоторного развития, однако, в ряде случаев они возникают с рождения или даже во внутриутробном периоде.
У пациентов с моногенными синдромами, сопровождающимися судорогами, как правило, определяется специфический симптомокомплекс, при котором судороги являются одним из его симптомов.
Структурные эпилепсии
Подтверждённой структурной причиной эпилепсии считают изменения головного мозга, которые могут быть выявлены с помощью методов нейровизуализации и которые в совокупности с клиническими и нейрофизиологическими данными позволяют с высокой долей вероятности предположить их связь с возникновением эпилептических приступов [5, 6]. Связанные с эпилепсией структурные изменения могут быть приобретёнными (например, вследствие черепно-мозговой травмы или внутриутробной инфекции) или генетически обусловленными (например, нарушения развития коры). У отдельных пациентов возможно сочетание различных потенциально эпилептогенных структурных изменений головного мозга (например, склероза гиппокампа и фокальной кортикальной дисплазии).
Церебральные дизонтогенезии, или мальформации развития коры, по современным данным, являются наиболее частой причиной эпилепсии, особенно проявляющейся у детей и подростков, и представляют собой большой спектр нарушений, поражающий целиком обе гемисферы (например, лиссэнцефалия), либо распространенные билатеральные поражения (например, билатеральная узловая гетеротопия), либо целиком одну гемисферу (например, гемимегалэнцефалия), либо изолированные участки коры одного полушария (фокальные кортикальные дисгенезии). Тяжелые дизонтогенетические поражения мозга, проявляющиеся эпилепсией и слабоумием, были идентифицированы в начале прошлого столетия на основании патологоанатомических исследований [Alzheimer A., 1907]. Клинико-морфологические сопоставления позволили описать клиническую картину многих таких синдромов и диагностировать их в младенчестве и раннем детстве. Это относится прежде всего к болезни Штурге - Вебера, лиссэнцефалии, туберозному склерозу, гемимегалэнцефалии. В диагностике ряда других дисплазий, лежащих в основе эпилепсии, особенно фокальной корковой дисплазии, как и ряда гетеротопий, выдающуюся роль сыграла магнитно-резонансная томография (МРТ) [6]. Классификация нарушений кортикального развития представлена в таблице 1 [65].
Таблица 1. Классификация нарушений кортикального развития [525]
Группа I. Нарушения вследствие аномальной пролиферации нейронов и глии или апоптоза | |
|---|---|
I. A | Микроцефалия |
I.B | Мегалэнцефалии, включая гемимегалэнцефалию |
I.C | Кортикальные дисгенезии с аномальной клеточной пролиферацией (ФКД II типа по классификации МПЭЛ, 2011). |
Группа II. Нарушения вследствие аномальной нейрональной миграции | |
II. A | Гетеротопия серого вещества |
II. B | Лиссэнцефалия |
II. C | Подкорковая ленточная гетеротопия (агирия – пахигирия – ленточный спектр) |
II. D | «Булыжниковая» мальформация |
Группа III. Нарушения вследствие аномалии постмиграционного развития (аномалии корковой организации) | |
III. A | Полимикрогирия |
III. B | Шизэнцефалия |
III. C | Фокальные кортикальные дисплазии (ФКД I и III типов по класс. МПЭЛ, 2011). |
Фокальная кортикальная дисплазия (ФКД) представляет собой участок мальформации коры головного мозга, который может иметь различные размеры и локализацию. Локальные изменения коры могут проявляться изменениями самих клеток коры (например, цитомегалия нейронов, баллонные клетки), аномальным их расположением (гетеротопии нейронов в слоях неокортекса или в подкорковом белом веществе), а также дезорганизацией коры и полимикрогирией. Эксперты МПЭЛ выделяют три типа ФКД [10, 11].
Тип I характеризуется в первую очередь аномальной ламинацией неокортекса в виде: нарушения радиальной миграции клеток с образованием «микроколонн» нейронов (тип Ia), нарушения шестислойного строения коры и нечёткости границы серого и белого вещества (тип Ib) или их сочетанием (тип Ic). Для ФКД I типа не характерно наличие морфологически изменённых клеток, но могут присутствовать незрелые клетки малого диаметра или гипертрофические пирамидные клетки с нормальной морфологией вне 5 слоя коры. Поскольку плотность серого вещества значимо не изменяется, ФКД I типа в большинстве случаев не удаётся выявить методами нейровизуализации.
Тип II характеризуется, помимо грубого нарушения послойного строения коры, наличием дисморфических нейронов большого диаметра. ФКД II типа дополнительно разделяют на подтипы в зависимости от отсутствия (тип IIa) или наличия (тип IIb) баллонных клеток. ФКД II типа, особенно IIb типа, чаще можно выявить с помощью магнитно-резонансной томографии по таким признакам как локальное утолщение коры, нечёткость границы серого и белого вещества, изменение сигнала от серого и подкоркового белого вещества, а также нарушения строения борозд и извилин.
Тип III определяется как сочетание нарушения ламинации коры и иных значимых структурных изменений той же или соседней области головного мозга, а именно: склероза гиппокампа (тип IIIa), опухоли (тип IIIb), сосудистой мальформации (тип IIIc), других структурных изменений (тип IIId).
ФКД часто ассоциирована с наличием у пациента эпилептических приступов и сопровождается, особенно ФКД II типа, интериктальной эпилептиформной активностью на ЭЭГ, совпадающей по локализации с областью структурных изменений коры [11]. ФКД, как правило, не приводит к клинически значимому неврологическому дефициту, проявляясь исключительно эпилептическими приступами, семиология которых зависит от локализации поражения. Приступы могут дебютировать в любом возрасте и зачастую резистентны к медикаментозной терапии [10].
Склероз гиппокампа является самым частым структурным изменением головного мозга у пациентов с фармакорезистентной эпилепсией [7]. На МРТ склероз гиппокампа характеризуется уменьшением объёма гиппокампа, усилением сигнала на T2-взвешенных изображениях, а также нарушением своей внутренней архитектуры [8]. Патоморфологически эксперты МПЭЛ выделяют три типа склероза гиппокампа в зависимости от вовлечения различных анатомических сегментов гиппокампа: наиболее часто встречаемый тип 1 предполагает наличие склероза в той или иной степени во всех сегментах (от СА1 до СА4), тип 2 – преимущественное вовлечение в патологический процесс сегмента СА1 и тип 3 – преимущественное поражение СА4 сегмента [9]. В отдельный тип выделяют изменения гиппокампа по типу глиоза без патогистологических признаков склероза (т.е. без уменьшения числа нейронов), однако его клиническая значимость остаётся объектом изучения [9]. Склеротические изменения нервной ткани могут распространяться на соседние структуры за пределами гиппокампа, например, на миндалевидное тело и парагиппокампальную извилину [8].
Склероз гиппокампа зачастую сопровождается интериктальной эпилептиформной активностью на ЭЭГ, исходящей из данной области. Оперативное вмешательство на височной доле у пациентов со склерозом гиппокампа в 2/3 случаев приводит к освобождению от приступов с нарушением сознания как минимум в течение года [8].
Тем не менее, открытым остаётся вопрос, является ли склероз гиппокампа непосредственной причиной или следствием иного этиологического фактора эпилепсии. Так, склероз гиппокампа может сопровождаться другими структурными изменениями головного мозга, такими как фокальная кортикальная дисплазия, высокодифференцированные опухоли или сосудистые мальформации [8]. Известно также, что гиппокамп может претерпевать структурные изменения в результате продолжительной эпилептической активности во время эпилептического статуса, а также после черепно-мозговой травмы или воспаления [7].
Опухоли головного мозга могут являться причиной развития судорожных приступов при условии сдавления или вовлечения в патологический процесс коры головного мозга. Примерами могут служить менингиома или диффузная инфильтративно растущая глиома. В отдельную группу выделяют доброкачественные опухоли, ассоциированные с длительно присутствующей, фармакорезистентной эпилепсией – «long-term epilepsy associated tumors» (LEATs): к ним относятся в первую очередь ганглиоглиома и дисэмбриопластическая нейроэпителиальная опухоль, а также более редкие варианты, такие как, например, ангиоцентрическая глиома, изоморфная диффузная глиома, папиллярная глионейрональная опухоль [12,13]. Для группы LEATs характерны дебют эпилепсии в молодом возрасте (чаще до 13 лет) и височно-долевая локализация опухоли в большинстве случаев [12,13]. Появление опухолей группы LEATs связывают с нарушением развития мозга на ранних этапах, что объясняет их частую ассоциацию с кортикальными дисплазиями [13]. Остаётся неясным, что именно является причиной стойкой предрасположенности к возникновению эпилептических приступов и резистентности к противоэпилептическим препаратам – непосредственно опухоль или изменения прилежащей мозговой ткани [12].
Черепно-мозговая травма является наиболее частой причиной (фактором риска) приобретённой структурной эпилепсии. Приступы, возникшие в течение первых 24 часов после травмы, называют немедленными, а в течение 2 - 7 суток – ранними; они являются острыми симптоматическими приступами, спровоцированными травмой. Приступы, возникшие после 7 суток, называют поздними, их считают неспровоцированными эпилептическими приступами, т.е. проявлением эпилепсии [14,15].
Вероятность развития эпилепсии после черепно-мозговой травмы, по данным разных исследований, варьирует от 5 до 42% в зависимости от выборки и времени наблюдения [14]. Посттравматическая эпилепсия более чем в 90% случаев развивается в течение первых двух лет [15]. По прошествии 5 лет риск развития заболевания значительно снижается (< 1%), но вероятность развития неспровоцированных приступов сохраняется в течение 10 и даже 30 лет после черепно-мозговой травмы [14].
Факторами риска развития посттравматической эпилепсии являются тяжёлая черепно-мозговая травма, множественные ушибы головного мозга, повреждение твёрдой мозговой оболочки, вдавленный перелом черепа, внутричерепное кровоизлияние, а также длительность потери сознания или амнезия более суток. Наличие ранних посттравматических приступов также может увеличивать риск развития эпилепсии [15].
Медикаментозная противоэпилептическая терапия может быть эффективна, но не во всех случаях удаётся достичь ремиссии [14]. Часто, примерно в 1/3 случаев посттравматической эпилепсии, на магнитно-резонансной томографии кроме посттравматических изменений выявляется также склероз гиппокампа [15].
Перинатальные поражения ЦНС (антенатальные, натальные и ранние постнатальные) являются частыми этиологическими факторами развития эпилепсии у детей. Гипоксически-ишемические поражения ЦНС вследствие перинатальных факторов включают: внутриутробные инфекции, перинатальные инсульты, паренхиматозные кровоизлияния, билирубиновую энцефалопатию, поствакцинальные поражения ЦНС, наследственные болезни метаболизма [65].
Перинатальные поражения – наиболее частая причина неонатальных судорог, которые связаны главным образом с асфиксией плода (развитием гипоксически-ишемической энцефалопатии) и механической травмой головного мозга, нередко сопровождающихся внутричерепными кровоизлияниями [6].
Инсульт и его последствия являются одной из основных причин (факторов риска) эпилепсии среди лиц старшего возраста. В зависимости от времени, прошедшего с момента инсульта, приступы разделяют на ранние, возникшие в течение первых 7 суток, и поздние, возникшие после 7 суток. Ранними считаются острые симптоматические приступы, спровоцированные локальными метаболическими изменениями и потому не являющиеся непосредственным проявлением эпилепсии. При этом, наличие ранних приступов увеличивает риск развития эпилепсии у пациента в дальнейшем. Поздние приступы, наоборот, считают проявлением приобретённой предрасположенности головного мозга к возникновению эпилептических приступов, т.е. проявлением постинсультной эпилепсии [15,16].
Распространённость постинсультной эпилепсии достигает 12 - 15%, по данным разных исследований, но различается в зависимости от методологии исследования и длительности наблюдения [16,17]. Помимо наличия ранних приступов, факторами риска развития постинсультной эпилепсии являются: возраст до 65 лет, гипонатриемия, злоупотребление алкоголем в анамнезе, геморрагический тип инсульта, вовлечение коркового вещества, височнодолевая локализация поражения, а также тяжёлый неврологический дефицит в дебюте инсульта [16,18]. Фокальная эпилептиформная активность на ЭЭГ также является прогностически неблагоприятным фактором развития постинсультной эпилепсии [15].
Эпилептические приступы, ассоциированные с инсультом, в большинстве случаев поддаются медикаментозному контролю, однако до 25% пациентов не достигают ремиссии [15]. Эффективность профилактического назначения противоэпилептических препаратов в настоящее время не доказана [16].
Метаболические эпилепсии
Неонатальные судороги могут возникать при различных врожденных нарушениях метаболизма: органических ацидуриях, аминоацидопатиях, дефектах ферментов дыхательной цепи, расстройствах метаболизма пирувата, нарушениях обмена ẞ - окисления жирных кислот, расстройствах метаболизма карнитина и др. Многие метаболические причины развития эпилепсии также обусловлены генетически. В таблице 2 представлены курабельные наследственные метаболические заболевания, которые нельзя пропустить [65].
Таблица 2. Наследственные метаболические заболевания [526, с добавлением]
1. | Дефицит В6 и фолиевой кислоты |
|---|---|
2. | Дефицит транспорта глюкозы, тип I (болезнь Де Виво) |
3. | Синдром гиперинсулинизма с аммониемией |
4. | DEND (задержка развития, эпилепсия, неонатальный диабет) |
5. | Гиперэкплексия |
6. | Нарушение синтеза креатинина |
7. | Дефицит биосинтетазы серина |
8. | Биотинидазная недостаточность |
9. | Дефицит фолата мозга |
10. | Нарушение синтеза биоптерина |
11. | Дефицит орнитинтранскарбамилазы (нарушение цикла мочевой кислоты) |
Инфекционные эпилепсии
Под инфекционной этиологией эпилепсии понимают известную инфекцию, ключевым проявлением которой являются приступы. Эпилепсия в данном случае возникает вследствие нейроинфекции и характеризуется формированием стойкой предрасположенности мозга к возникновению приступов, а не только судорожными приступами в остром периоде инфекционного заболевания. Обусловленные нейроинфекцией изменения головного мозга также могут быть структурными [5].
Примерами нейроинфекций, способных привести к развитию эпилепсии, являются клещевой энцефалит, вирус Зика, цитомегаловирусная инфекция, вирус иммунодефицита человека, туберкулёз. К инфекционным эпилепсиям относят также эпилепсии, развивающиеся при инвазионных заболеваниях, например, при цистицеркозе, токсоплазмозе, эхинококкозе [5].
Иммунные эпилепсии. Причиной иммунной эпилепсии считают иммунное расстройство, основным проявлением которого являются приступы, и которое непосредственно приводит к развитию эпилепсии [5]. В большинстве случаев данным иммунным расстройством является аутоиммунный процесс, триггером которого служат онкологическое заболевание или инфекция, в том числе вирусный энцефалит [19]. Приступы, возникающие в результате аутоиммунного энцефалита, зачастую могут быть первым, преобладающим или даже единственным его проявлением и возникают с частотой от 33% до 100% случаев в зависимости от антигена [20]. Однако, далеко не всегда аутоиммунный энцефалит приводит к развитию эпилепсии как хронического заболевания, и часто приступы прекращаются после завершения острого периода болезни, который может длиться несколько месяцев. Таким образом, диагноз эпилепсии рекомендуют подтверждать после длительного наблюдения пациента с продолжающимися приступами, например, в течение 12 месяцев [20].
Различают аутоиммунные энцефалиты, характеризующиеся наличием антител: 1) к поверхностным клеточным антигенам, 2) к внутриклеточным антигенам (опосредован Т-клеточным иммунитетом). В первом случае риск формирования стойкой предрасположенности к возникновению эпилептических приступов после разрешения энцефалита, как правило, низкий, за исключением анти-LGI1 и анти-GABAaR энцефалитов. Во втором случае вероятность развития эпилепсии в исходе энцефалита, напротив, высокая. Примерами энцефалитов с антителами к внутриклеточным антигенам являются некоторые паранеопластические энцефалиты (анти-Hu, анти-Yo, анти-CRMP-5) и анти-GAD65 энцефалит [19,20].
Однако, несмотря на выдающиеся достижения в диагностике этиологии эпилепсии (прежде всего – высокоразрешающие методы нейровизуализации и генетического тестирования), большое количество форм проходят под рубрикой «неизвестная причина» (по старой терминологии «криптогенная эпилепсия»). Процент неустановленной этиологии колеблется в широком диапазоне от 20% до 64% всех случаев. По данным клинико-эпидемиологического исследования в РФ, фокальная эпилепсия неуточненной этиологии встречалась примерно в 34% случаев [45]. Процент выявляемости этиологических факторов эпилепсии у детей достоверно выше, чем у взрослых, несмотря на большее их разнообразие [527].
1.2.2. Патогенез эпилепсии
Патогенез эпилепсии не является единым для всех форм заболевания, хотя имеются общие универсальные звенья. Патогенез эпилепсии любого типа включает процесс эпилептогенеза: постепенное развитие судорожной активности и стадию сформировавшейся эпилепсии, причем эпилептогенез может продолжаться и при развившейся эпилепсии (рис. 1).
Рис. 1. Периодизация основных блоков патогенеза приобретенной эпилепсии
Общим признаком, характерным для патогенеза всех форм эпилепсии, является судорожная активность, вызванная пароксизмальными разрядами групп нейронов в результате избыточного возбуждения и/или недостаточного торможения. Возникшая в определенном участке мозга избыточная электрическая активность распространяется в соседние зоны, а также может передаваться к мышцам, вызывая конвульсии. Как правило, распространение возбуждения в подкорковые, таламические, стволовые и спинальные структуры соответствует тонической фазе судорожного приступа, а последующий тормозной импульс из таламуса прерывает тоническую фазу, которая сменяется спорадическими вспышками электрической активности в клонической фазе. В настоящее время единственным приемлемым биомаркером эпилептогенеза следует признать патологические высокочастотные осцилляции [24].
В формирование судорожной активности вносят вклад возбуждающие и тормозные постсинаптические потенциалы, изменения потенциалзависимых ионных каналов, а также изменения локальных концентраций ионов. Основным возбуждающим нейромедиатором является глутамат, реализующий свое действие через 2 типа глутаматных рецепторов: ионотропные, опосредующие быструю синаптическую трансмиссию (глутаматзависимые АМРА-, каинатные и NMDA-ионные каналы), и метаботропные, опосредующих медленную синаптическую трансмиссию (связаны с G-белками и регуляцией вторичных посредников цAMФ и фосфолипазы C). Основным тормозным нейромедиатором в ЦНС является гамма-аминомасляная кислота (ГАМК), имеющая в ЦНС 2 типа рецепторов: GABAA, постсинаптические специфичные, сопряженные с CI- -каналами и GABAB, пресинаптические ауторецепторы, снижающие высвобождение медиатора за счет снижения притока Са++ и сопряженные с постсинаптическими G-белками, что способствует повышению тока K+.
Клеточные механизмы генерации избыточного возбуждения при судорожной активности на уровне ионного равновесия включают токи Na+ и Ca++ в клетки за счет избытка нейромедиаторов глутамата и аспартата и изменения свойств их рецепторов. Недостаточное торможение за счет потока CI- в клетку и K+ из клетки обусловлено недостатком ГАМК или изменением свойств соответствующих рецепторов. К нейронным факторам, регулирующим возбудимость нейронов, относятся тип ионных каналов, их число и распределение на мембране нейрона, посттрансляционные модификации каналов (напр. фосфорилирование), активация систем вторичных посредников, влияющих на функционирование каналов (напр. G-белков), модуляция экспрессии генов ионных каналов. Некоторые формы эпилепсии тесно связаны с каналопатиями, являющимися результатом наследственных мутаций лиганд-зависимых ионных каналов (т. е. ионотропных рецепторов) и потенциалзависимых ионных каналов. Выявлены также типы приобретенной аутоиммунной эпилепсии, связанной с образованием антител против калиевых, натриевых и хлоридных каналов, а также изменением экспрессии каналов после судорог. При этом различные мутации одного и того же гена могут вызывать совершенно разные типы судорог и эпилепсии [23]. К синаптическим факторам, модифицирующим возбудимость нейронов, относятся изменения экспрессии лигандзависимых ионотропных каналов, посттрансляционные изменения в таких каналах, ремоделирование локализации или конфигурации синапсов, изменение синаптических функций щелевых контактов. Несинаптические (внешние) факторы модификации возбудимости нейронов включают изменения внеклеточных концентраций ионов, изменения внеклеточного пространства, модуляцию метаболизма нейромедиаторов или их захвата клетками глии. Механизмы генерации гипервозбудимости на сетевом уровне включают аксональный спрутинг возбуждающих нейронов, потерю тормозных нейронов, потерю возбуждающих нейронов, осуществляющих контроль тормозных нейронов, изменения импульсной активности нейронов (например при каналопатиях).
К общим признакам патогенеза эпилепсии относится воспаление, которое может быть связано с инфекцией или обусловлено нарушениями иммунной системы. Связь нейровоспаления и патогенеза эпилепсии, в т. ч. его важная роль в период эпилептогенеза, прослежена при различных формах эпилепсии в клинике и эксперименте [25]. Считается, что хронизации воспалительного процесса при эпилепсии способствуют активация микроглии и астроглиоз, сопровождающиеся повреждением нейронов. Одним из триггеров воспаления в ЦНС является повреждение гематоэнцефалического барьера, гематоликворного и ликвороэнцефалического барьеров, а также относительно автономного иммунного барьера мозга. В нейровоспалении и противовоспалительной защите мозга принимает участие система провоспалительных и противовоспалительных цитокинов, которые могут обладать как про-, так и антиконвульсантной активностью. Дисбаланс цитокиновой системы с преобладанием провоспалительных компонентов (интерлейкина 6, интерлейкина 1β, фактора некроза α) описан как у пациентов с эпилепсией, так и в различных моделях на животных и связан в первую очередь с активацией микроглии, а затем астроцитов. Активация цитокиновой системы как в количественном, так и в качественном отношении зависит от типа и выраженности судорожной активности, а также периода эпилепсии. Провоспалительные цитокины могут быть вовлечены в развитие гиперсинхронности нейронов и гипервозбудимости головного мозга за счет разных механизмов, в том числе взаимодействия с возбуждающими и тормозными нейромедиаторными системами, системой оксида азота в глиальных клетках и нейронах и сигнальными каскадами, приводящими к нейродегенерации и гибели нейронов. Некоторые из параметров магнитно-резонансной томографии коррелируют с наличием патологических высокочастотных осцилляций, могут косвенно отражать текущий воспалительный процесс в головном мозге и стать возможными биомаркерами эпилептогенеза. Общими патогенетическими механизмами развития эпилепсии, наряду с нейровоспалением и сопутствующим глиозом, являются также дисбаланс активных форм кислорода и окислительный стресс, нарушения систем антиоксидантной защиты (например, системы глутатиона). Дисфункция митохондрий сопровождается избыточной генерацией супероксидного анион-радикала и развитием окислительного повреждения ключевых молекул и клеточных органелл на фоне дефицита энергетических субстратов, в первую очередь аденозинтрифосфата (АТФ).
Медиальная височная эпилепсия (МВЭ), наиболее часто встречающаяся форма эпилепсии, подразумевает эпилептический синдром, при котором судорожная активность возникает из височной доли при активном вовлечении гиппокампа. Именно на МВЭ сфокусирована существенная часть фундаментальных исследований патогенеза эпилепсии, которые включают в первую очередь изучение патологии и патофизиологии гиппокампа при эпилептогенезе [26]. Возможные механизмы отложенного эпилептогенеза активно обсуждаются в рамках нескольких гипотез. Одна из них, модель киндлинга, предполагает, что повторяющиеся субконвульсивные стимулы, приводящие к последующим электрическим разрядам (afterdischarges), могут в конце концов приводить к развитию спонтанных судорог (эпилепсии). Другая модель рассматривает патофизиологию и изменения сетей гиппокампа при эпилептогенезе. Эта модель непосредственно связана с эпилепсией височной доли, но может быть полезна и для понимания развития других типов эпилепсии. Следует учитывать, что гиппокамп, а именно его зубчатая извилина, является еще и нейрогенной нишей, в которой образование новых нейронов (нейрогенез) продолжается в течение всей жизни. Зубчатая извилина выполняет функцию привратника (фильтра); в норме тормозная иннервация гранулярных клеток доминирует над возбуждающей, что позволяет зубчатой извилине контролировать возбуждение, при этом иннервация тормозных ГАМК-ергических интернейронов гранулярными клетками по механизму отрицательной обратной связи контролирует возбудимость гиппокампа.
Установлено, что при эпилептогенезе существенно усилено образование новых нейронов в зубчатой извилине гиппокампа. Эпилептиформная активность возникает, когда зубчатая извилина не может выполнить свою функцию фильтра возбуждения. Причиной этого может быть формирование аберрантных нервных сетей за счет вызванной нарушенным нейрогенезом реорганизации связей между гранулярными клетками. Предполагается, что именно формирование рекуррентных возбуждающих связей в процессе эпилептогенеза нарушает функцию зубчатой извилины. При этом аномальная интеграция новорожденных гранулярных клеток в гиппокампальные сети происходит за счет прорастания аксонов мшистых волокон, прорастания базальных дендритов в хилус, где они образуют синаптические контакты с мшистыми волокнами, миграции гранулярных клеток в хилус с нарушением морфологии гранулярного слоя. Эти изменения нарушают функционирование гиппокампа, способствуя существованию проэпилептогенных нервных сетей. Нейродегенерация и снижение нейрогенеза гиппокампа являются одним из общих патогенетических механизмов, лежащих в основе эпилептогенеза. Как и при ряде других неврологических и психических заболеваний, эти события тесно связаны с дисрегуляцией нейротрофинов, в частности, нейротрофического фактора мозга (с англ. Brain Derived Neurotrophic Factor, BDNF) [545]. Предполагается, что при лимбическом эпилептогенезе усиленная экспрессия BDNF вносит ключевой вклад в аберрантный нейрогенез и спрутинг мшистых волокон гиппокампа, способствуя тем самым длительной потенциации возбуждающей синаптической трансмиссии [27, 28]. Важно отметить, что на долю МВЭ приходится существенная часть пациентов с фармакорезистентной эпилепсией, при этом фармакорезистентность является проявлением как общей тяжести заболевания, так и результатом глубокого дисбаланса между многокомпонентной эпилептической и противоэпилептической системами головного мозга. Возможно, в основе фармакорезистентности лежат врождeнные или приобретeнные изменения активности белков-транспортеров гематоэнцефалического барьера и/или чувствительность молекулярных мишеней противоэпилептических препаратов.
У пациентов с опухолями мозга за счет снижения перфузии в области опухоли и усиления метаболизма возникает гипоксия, вызывающая ацидоз и нарушения окислительного энергетического метаболизма, что приводит к набуханию клеток глии и повреждению окружающей ткани. Возникающий дисбаланс между возбуждением и торможением приводит к судорожной активности за счет повышенного внеклеточного уровня глутамата до нейротоксических значений. При глиоме эпилептическая активность возникает вне опухоли в районе околоопухолевой границы, где повышен уровень глутамата. Активность рецепторов ГАМК понижена, что также вносит вклад в развитие избыточного возбуждения [29].
Механизмы эпилептогенеза в результате ЧМТ сводятся к вопросу о причинах развития склонности к повторяющимся неспровоцированным судорожным приступам при отсутствии явных патологических провоцирующих факторов в позднем периоде ЧМТ. Первичные повреждения при ЧМТ включают в себя острую клеточную гибель, нарушение гематоэнцефалического барьера; они приводят к деполяризации нейронов, выбросу возбуждающих нейромедиаторов и повышению экстраклеточной концентрации К+, а в конечном итоге к гиперсинхронизации нейронов, что проявляется острыми судорожными приступами у животных и, вероятно, у человека [30]. К клеточной гибели приводит острый чрезмерный выброс глутамата и аспартата, вызывающий активацию NMDA-рецепторов, вход Na+ и Ca++ в клетку, выбросу K+, апоптоз и некроз нейронов в результате эксайтотоксичности[31]. Вторичное повреждение в результате ЧМТ связано с активацией процессов отложенной клеточной гибели, нейровоспаления, глио- и ангиогенеза. Многие из этих процессов вовлечены в эпилептогенез: гибель нейронов, глиоз, нейровоспаление, нарушение гематоэнцефалического барьера, нарушение возбудимости нейронов, нарушенные ангиогенез и нейрогенез, изменение синаптической пластичности, перестройка нейрональных сетей, изменения экспрессии генов и эпигенетические модификации [32]. Помимо формирования прямого очага повреждения в коре, ЧМТ приводит к дистантной и вторичной гибели нейронов и активации глии в гиппокампе [33], в первую очередь ГАМК-ергических вставочных нейронов хилуса [34]. Нейровоспаление после ЧМТ присутствует как в остром периоде, обусловливая отёк и нейродегенерацию, так и в хроническом периоде [35]. Клеточный субстрат нейровоспаления в остром периоде в основном представлен микроглией, в хроническом большую роль играют астроциты. Синтезируемые иммунными клетками цитокины модифицируют функцию глутамат- и ГАМК-ергических рецепторов, ингибируют захват глутамата астроцитами, нарушают функцию потенциал-зависимых ионных каналов, ведут к повышению экстраклеточной концентрации К+, и всё это формирует основу для нейронной гиперсинхронизации [25]. Дальнейшее прогрессирующие изменения в гиппокампе тесно связаны с изменениями нейропластичности: наблюдается спрутинг мшистых волокон в зубчатой извилине гиппокампа формируются новые возбуждающие синапсы на гранулярных клетках, что способствует большей дивергенции возбуждения в гиппокампе, а также самоактивации гранулярных клеток [36, 37]. В результате длительно протекающих структурных и метаболических изменений в гиппокампе в отдалённом периоде ЧМТ наблюдаются прогрессирующие по частоте и выраженности эпилептические приступы на фоне снижении порога возбудимости нейронов[38-40].
1.3 Эпидемиология заболевания или состояния (группы заболеваний или состояний)
1.3.1. Эпидемиология эпилепсии
Более 50 миллионов человек во всем мире страдают эпилепсией [41]. На эпилепсию приходится 13 миллионов лет жизни, скорректированных на инвалидность [42].
Согласно определению Международной противоэпилептической Лиги, наличие активной эпилепсии у пациента подразумевает прием противоэпилептических препаратов либо наличие приступов за последние 2 - 5 лет [43]. Стандартизированная по возрасту распространенность активной эпилепсии в мире по данным на 2016 год составляет 621,5 (540,1 – 737,0) на 100000. Распространенность активной эпилепсии увеличивается с возрастом, достигая максимума к 5 - 9 годам (374,8 [280,1 – 490,0]) и у людей старше 80 лет (545,1 [444,2 – 652,0])[42]. Заболеваемость эпилепсией в разных странах составляет в среднем 67.77 на 100000 человек в год (95% CI 56,69 – 81,03) [44].
По данным единственного российского масштабного клинико-эпидемиологического исследования 517624 человек 14 лет и старше в 14 регионах РФ (0,34% всего населения РФ) стандартизированное по возрасту значение распространенности (European Standard Million) составило 3,40 случая на 1000. Распространенность эпилепсии была выше: в Сибири и на Дальнем Востоке по сравнению с Европейской частью РФ, в сельской местности по сравнению с крупными городами. Возрастная структура заболеваемости отличалась от наблюдаемой в странах Европы и США – значения заболеваемости были ниже в старших возрастных группах[45].
Каждый год регистрируется 125000 смертей больных эпилепсией [46]. Стандартизированные показатели смертности пациентов с эпилепсией в странах с низким и средним уровнем дохода более чем в 2,5 раза, а в странах с высоким уровня дохода – в 2 - 7 раз - превышают общепопуляционные [47,48].
Преждевременная смертность больных эпилепсией обусловлена в том числе более частой травматизацией и суицидами, а также высоким уровнем соматической и психиатрической коморбидности [49–51].
Особое место среди причин смерти пациентов с эпилепсией занимает SUDEP (sudden unexpected death in epilepsy), или синдром внезапной смерти при эпилепсии, так как частота встречаемости этого синдрома среди молодых людей с эпилепсией, в особенности фармакорезистентной, по разным оценкам в 24 - 27 раз выше, чем в общей популяции [52].
1.3.2. Эпидемиология эпилептического статуса
Критический анализ популяционных исследований в различных регионах земного шара показал высокую вариабельность возникновения эпилептического статуса от 1,29 до 73,7 на 100000 взрослых [53]. По данным большинства исследований, риск развития эпилептического статуса выше у мужчин в сравнении с женщинами, а также у детей и людей старше 60 лет [54].
1.4 Особенности кодирования заболевания или состояния (группы заболеваний или состояний) по Международной статической класификации болезней и проблем, связанных со здоровьем
Согласно Международной классификации болезней 10-го пересмотра (МКБ-10), к эпилепсии относятся преимущественно коды G40 и G41 [55]:
•G40.0. Локализованная (фокальная, парциальная) идиопатическая эпилепсия и эпилептические синдромы с судорожными приступами с фокальным началом. Доброкачественная детская эпилепсия с пиками на ЭЭГ в центрально-височной области. Детская эпилепсия с пароксизмальной активностью на ЭЭГ в затылочной области;
•G40.1. Локализованная (фокальная, парциальная) структурная эпилепсия и эпилептические синдромы с простыми парциальными приступами.
Приступы без изменения сознания. Простые парциальные приступы, переходящие во вторично-генерализованные приступы;
•G40.2. Локализованная (фокальная, парциальная) структурная эпилепсия и эпилептические синдромы со сложными парциальными приступами.
Приступы с изменением сознания, часто с эпилептическими автоматизмами. Сложные парциальные приступы, переходящие во вторично-генерализованные приступы;
•G40.3. Генерализованная идиопатическая эпилепсия и эпилептические синдромы. Доброкачественная миоклоническая эпилепсия раннего детского возраста. Неонатальные судороги(семейные). Детские абсансы (пикнолепсия). Эпилепсия с большими судорожными приступами (grand mal) при пробуждении. Юношеская абсансная эпилепсия, миоклоническая эпилепсия (импульсивный малый приступ (petit mal)). Неспецифические эпилептические приступы: атонические, клонические, миоклонические, тонические, тонико-клонические;
•G40.4. Другие виды генерализованной эпилепсии и эпилептических синдромов. Эпилепсия с миоклоническими абсансами, миоклонико-атоническими приступами. Синдром Леннокса – Гасто. Структурная ранняя миоклоническая энцефалопатия. Синдром Веста;
· G40.5. Особые эпилептические синдромы.
Эпилепсия парциальная непрерывная (Кожевникова). Эпилептические приступы, связанные с употреблением алкоголя, применением лекарственных средств, гормональными изменениями, лишением сна, воздействием стрессовых факторов. При необходимости идентифицировать лекарственное средство используют дополнительный код внешних причин (класс ХХ);
•G40.6. Приступы grand mal неуточненные (с приступами petit mal или без них);
•G40.7. Приступы petit mal неуточненные без приступов grand mal;
•G40.8. Другие уточненные формы эпилепсии.
Эпилепсия и эпилептические синдромы, не определенные как фокальные или генерализованные;
•G40.9. Эпилепсия неуточненная;
•G41.0. Эпилептический статус grand mal;
•G41.1. Эпилептический статус petit mal;
•G41.2. Сложный парциальный эпилептический статус;
•G41.8. Другой уточненный эпилептический статус;
•G41.9. Эпилептический статус неуточненный;
•G83.8. Паралич Тодда;
•F80.3. Синдром Ландау – Клеффнера (приобретенная эпилептическая афазия);
•R56.0. Судороги при лихорадке;
•R56.8. Другие и неуточненные судороги;
•P90. Неонатальные судороги (исключено: семейные неонатальные судороги – G40.3).
1.5 Классификация заболевания или состояния (группы заболеваний или состояний)
Новая классификация эпилепсий МПЭЛ 2017 г. является многоуровневой и предназначена для применения в клинической практике (см. рис. 2) [5].
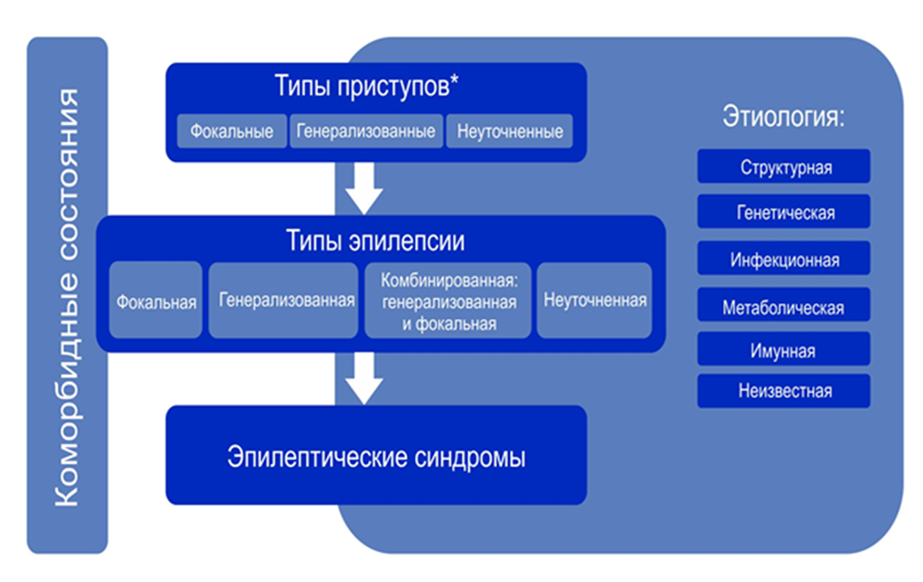
Рис.2. Схема классификации эпилепсий МПЭЛ 2017 г.
Данная классификация базируется на следующих принципах:
1. Определение типа приступов
2. Определение типа эпилепсии
3. Определение эпилептического синдрома
4. Определение этиологии эпилепсии
5. Определение коморбидных состояний
Классификация содержит несколько уровней, что обусловлено большой вариабельностью доступных методов обследования пациентов с эпилепсией в мире. На первом этапе (уровне) идет определение типа приступа: фокальный, генерализованный или с неизвестным началом (см. рис. 3). Фокальный эпилептический приступ определяется как приступ, исходящий из какой-либо области нейрональных сетей, ограниченных одним полушарием; эта зона может быть очень локальной или более распространенной. При этом возможно распространение на соседние зоны или переход на контралатеральное полушарие. Генерализованный эпилептический приступ определяется как приступ, исходящий из некоторой области головного мозга с быстрым распространением и билатеральным захватом нейрональных сетей. Неклассифицированный приступ определяется как приступ, который вследствие недостатка информации невозможно отнести к другим категориям в данный момент времени.

Рис. 6. Схема различных форм хирургического лечения в зависимости от этиологии эпилепсии, объема и локализации поражения, размера эпилептогенной зоны [376]. Использование интраоперационного нейрофизиологического мониторинга во время хирургического лечения фармакорезистентной эпилепсии. · Рекомендуется проведение интраоперационного нейрофизиологического мониторинга с картированием функций резецируемой коры головного мозга у пациентов с фармакорезистентной фокальной эпилепсией при расположении эпилептогенной зоны вблизи функционально значимой коры или подкорковых проводящих путей [409]. Уровень убедительности рекомендаций С (уровень достоверности доказательств – 4). Комментарий: Применение интраоперационного нейрофизиологического мониторирования (ИОМ) и картирования коры во время резекции эпилептогенной зоны позволяет локализовать функциональные зоны с помощью прямой электростимуляции коры. При расположении эпилептогенной зоны возле моторной коры, проведение электростимуляции возможно во время наркоза, а для проведения картирования речевых зон необходимо пробуждение пациента. У детей локализацию центральной борозды возможно проводить методом соматосенсорных вызванных потенциалов с анализом реверсии фазы над Роландовой бороздой. Нарушения психики различного спектра весьма частое явление при эпилепсии. · Рекомендуется консультация врача-психиатра для пациентов с эпилепсией при наличии у него коморбидных психических расстройств, поведенческих, эмоциональных, когнитивных нарушений [6]. Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5). · Рекомендуется назначение антидепрессантов из группы селективных ингибиторов обратного захвата серотонина (СИОЗС) для лечения депрессивного расстройства у ПЭ старше 18 лет [410, 411]. Уровень убедительности рекомендаций С (уровень достоверности доказательств – 3). Комментарий: Общие подходы к лечению депрессий у ПЭ аналогичны таковым у пациентов с депрессией без эпилепсии. Результаты проспективного открытого исследования показали, что на фоне приема СИОЗС 7 из 8 пациентов с эпилепсией и депрессией к 12 неделе терапии достигают ремиссии [411]. При недостаточной эффективности СИОЗС целесообразно рассмотреть вопрос о назначении антидепрессантов из других групп. · Рекомендуется назначение пароксетина** пациентам с эпилепсией 18 лет и старше при наличии депрессии [412, 413]. Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5). Комментарии: Результаты рандомизированного контролируемого сравнительного исследования пароксетина** в дозе 20 - 40 мг в сутки и других антидепрессантов, проведенного на 67 ПЭ и депрессией показали, что в течении 8 недель терапии, 82% достигли первичного ответа на лечение [412]. При этом из группы пароксетина** не выбыл ни один человек. · Рекомендуется назначение циталопрама пациентам с эпилепсией 18 лет и старше при наличии депрессии [414–416]. Уровень убедительности рекомендаций С (уровень достоверности доказательств – 4). Комментарии: В открытом проспективном исследовании 43 ПЭ и депрессией было показано, что на прием циталопрама в дозировке 10 - 40 мг в сутки в течении 8 недель ответило 65% ПЭ и депрессией [414]. В 16-недельном исследовании 45 ПЭ и депрессией похожего дизайна уровень ответа на терапию циталопрамом составил 86%. [415]. В проспективном открытом исследовании циталопрама, проведенного на пациентах с височной эпилепсией и депрессией, показано, что уровень ответа на терапию составил 53,3%, а 21,2% пациентов достигли ремиссии к 20 - 30 неделе терапии [416]. Ни в одном из приведённых выше исследований прием циталопрама не сопровождался учащением приступов. · Рекомендуется назначение сертралина** пациентам с эпилепсией старше 18 лет при наличии депрессии [417, 418]. Уровень убедительности рекомендаций С (уровень достоверности доказательств – 4). Комментарии: В рандомизированном исследовании сертралина** и когнитивно-поведенческой психотерапии было показано, что на фоне приема сертралина** в дозе 50 - 200 мг в сутки (N = 75) к 16 неделе терапии ремиссии достигли 53% пациентов [417]. Ни у одного пациента прием сертралина** не сопровождался учащением приступов. В течении 12 месяцев наблюдения за 100 пациентами с эпилепсией с депрессивными/обсессивными расстройствами (97/3) на фоне приема сертралина** лишь у 6 пациентов отмечалось учащение приступов, при этом причинную связь между приемом сертралина** и учащением приступов авторы смогли выявить лишь у одного пациента [418]. · Рекомендуется назначение венлафаксина для лечения депрессивного расстройства пациентам с эпилепсией 18 лет и старше [419, 420]. Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5). Комментарий: В результате рандомизированного контролируемого исследования было установлено, что в течении 8 недель терапии значимо большее число пациентов, получавших венлафаксин (25 - 75 мг в сутки) достигли первичного ответа на терапию по сравнению с теми, кто не получал какого-либо лечения депрессии (69% vs 19%). При этом авторами не проводилась оценка влияния венлафаксина на эпилептические приступы [419]. · Рекомендуется назначение миртазапина для лечения депрессивного расстройства у пациентов с эпилепсией 18 лет и старше [416, 420]. Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5). Комментарий: По данным проспективного открытого исследования назначение миртазапина в средней дозе 32,2 (6,8) мг в сутки привело к уменьшению симптомов депрессии у 51,9%, а около 15% достигли ремиссии к 20 - 30 неделе терапии [416]. Прием миртазапина не сопровождался учащением приступов. · Не рекомендуется назначение амитриптилина** для лечения депрессивного расстройства у ПЭ 18 лет и старше [421]. Уровень убедительности рекомендаций В (уровень достоверности доказательств – 2). Комментарий: В результате двойного слепого плацебо контролируемого исследования к 6 неделе терапии не было выявлено значимых различий по уровню ответа на 25 мг амитриптилина** и плацебо [421]. · Не рекомендуется назначение кломипрамина** для лечения депрессивного расстройства у ПЭ 18 лет и старше [422]. Уровень убедительности рекомендаций С (уровень достоверности доказательств – 4). Комментарий: результаты метанализа [422] показали, что кломипрамин** ассоциирован с повышенным риском эпилептических приступов у людей, не болеющих эпилепсией, со стандартизированным показателем заболеваемости 4 (95% CI 2,6 - 6,0). · Рекомендовано при назначении антидепрессантов пациентам с эпилепсией и депрессией 18 лет и старше учитывать их лекарственные взаимодействия с противоэпилептическими препаратами [410]. Уровень убедительности рекомендаций С (уровень достоверности доказательств – 3). Комментарии: карбамазепин**, фенитоин** и фенобарбитал** снижают концентрации СИОЗС примерно на 25% [423], а вальпроевая кислота** незначительно повышают концентрацию активного метаболита венлафаксина - О-десметилвенлафаксин [424]. Флувоксамин, флуоксетин** и в меньшей степени сертралин** незначительно повышают концентрацию вальпроевой кислоты** [425, 426]. Некоторым пациентам может потребоваться изменение режима дозирования ПЭП. · Рекомендуется взрослым пациентам c эпилепсией с симптомами тревоги проведение когнитивно-поведенческой психотерапии [427, 428]. Уровень убедительности рекомендаций С (уровень достоверности доказательств – 4). · Рекомендуется взрослым пациентам c эпилепсией и в зависимости от вида тревожных расстройств назначение антидепрессантов из группы СИОЗС (сертралин**, пароксетин**, флувоксамин, циталопрам, эсциталопрам, флуоксетин**) в качестве препаратов первой линии [430]. Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5). Комментарии: Терапия антидепрессантами из группы СИОЗС безопасна для пациентов с эпилепсией, так как они не влияют на частоту эпилептических приступов. Тем не менее, стоит учитывать возможные лекарственные взаимодействия с ПЭП, которые среди СИОЗС наиболее выражены у пароксетина**. Рекомендуемая суточная дозировка сертралина** – 50 - 150 мг в сутки, циталопрама и пароксетина** – 20 - 60 мг в сутки, эсциталопрама – 10 - 20 мг в сутки, флуоксетина** – 20 - 40 мг в сутки, флувоксамина – 100 — 300 мг в сутки. Обычно начинают с минимальных терапевтических доз, поднимая их при отсутствии эффекта. · Рекомендуется пациентам c эпилепсией старше 18 лет и генерализованным тревожным расстройством назначение прегабалина** [431]. Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5). Комментарии: механизм действия препарата заключается в связывании с альфа-2-дельта-субъединицей потенциалзависимых кальциевых каналов, что блокирует передачу болевых импульсов и снижает вероятность судорожных приступов, связывает пресинаптические N и P/Q кальциевые каналы, уменьшая их чрезмерную активность, таким образом снижая проявления тревожной симптоматики. Режим дозирования для лечения генерализованного тревожного расстройства: начальная доза — 150 мг/сут. В зависимости от достигнутого эффекта и переносимости, через одну неделю дозу можно увеличить до 300 мг/сут. Длительное применение препарата безопасно для пациента. Побочными эффектами прегабалина** являются такие явления, как: седация, головокружение, тремор, дизартрия, парестезии, нарушение памяти, нарушение координации, нарушение внимания, спутанность сознания, эйфория, раздражительность, сухость во рту, запор, увеличение веса, повышенный аппетит, метеоризм, помутнение зрения, диплопия, периферические отеки, снижение либидо, эректильная дисфункция. Паллиативная помощь – подход, целью которого является улучшение качества жизни пациентов и членов их семей, оказавшихся перед лицом угрожающего жизни заболевания. Эта цель достигается путем предупреждения и облегчения страданий благодаря раннему выявлению, тщательной оценке и купированию боли и других тягостных физических симптомов, а также оказанию психосоциальной и духовной поддержки [432]. Главная задача паллиативной помощи – достижение, поддержка, сохранение и повышение, насколько это возможно, качества жизни пациента. Однако определить, что такое «качество жизни», может только сам пациент, нуждающийся в паллиативной помощи [433]. · Рекомендуется оказание паллиативной медицинской помощи детям с формами эпилепсии, относящимися к заболеваниям, ограничивающим продолжительность жизни (коды МКБ G 40.4, G 40.5, G 40.9) [268–270] и детям с фармакорезистентной эпилепсией, которым выполнены паллиативные нейрохирургические вмешательства (каллозотомия и субпиальные транссекции) [271, 272], осуществляемой сотрудниками медицинских организаций и их подразделений, оказывающих специализированную паллиативную медицинскую помощь детям, для обеспечения потребностей пациента и его законных представителей. Уровень убедительности рекомендаций С (уровень достоверности доказательств - 5). · Рекомендуется введение защечного раствора мидазолама детям при эпилептическом статусе для купирования приступа судорог [434]. Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5). · Рекомендуется введение ректального раствора диазепама детям при эпилептическом статусе для купирования приступа судорог [434]. Уровень убедительности рекомендаций C (уровень достоверности доказательств 5). К наблюдению за детьми с формами эпилепсии, относящимися к заболеваниям, ограничивающим продолжительность жизни (коды МКБ G 40.4, G 40.5, G 40.9) и детям с фармакорезистентной эпилепсией, которым выполнены паллиативные нейрохирургические вмешательства (каллозотомия и субпиальные транссекции), должны привлекаться сотрудники медицинских организаций и их подразделений, оказывающих специализированную паллиативную медицинскую помощь детям. Паллиативная помощь детям с эпилепсией оказывается на основании соответствующих нормативных документов (см. Приложение «Связанные документы»). 3.5 Кетогенная диета Кетогенная диета (КД) - это хорошо зарекомендовавший себя нефармакологический метод лечения детей и взрослых с лекарственно-устойчивой эпилепсией, который основывается на изменении рациона с увеличением доли жиров и уменьшением углеводов и белков. Из-за очень высокой потребности в жирах классическую КД может быть трудно поддерживать, что приводит к необходимости использовать ее альтернативные варианты. Модифицированная КД, по-видимому, почти так же эффективна, но обеспечивает больший выбор продуктов питания. Различные альтернативы КД включают модифицированную диету Аткинса (МДА), диету со среднецепочечными триглицеридами (ДСЦТ) и терапию с низким гликемическим индексом (НУД или низкоуглеводная диета) [435]. Типы КД (1) Классическая КД состоит преимущественно из жиров (до 90%) и низкого содержания углеводов и белков, что приводит к кетозу и имитирует состояние голодания. Это жесткая диета, рассчитывается математически для каждого пациента индивидуально. Из-за ограничения рациона пациенту необходимо обеспечивать организм достаточным количеством витаминов и минералов. (2) МДА направлена на обеспечение разнообразия продуктов и вкусовых качеств с соотношением жиров к углеводам и белку 1 : 1 и содержит около 65% жира, 25% белка и 10% углеводов. Поощряется употребление жиров, а потребление углеводов ограничивается до 10 – 20 г/день для детей и 15 – 20 г/день для взрослых. МДА не требует взвешивания пищи на граммовых весах или ограничения калорий, белков или жидкостей и может быть хорошим вариантом для пациентов, которые не могут переносить более строгую диету, такую как классическая КД. При МДА рекомендуется прием поливитаминов с низким содержанием углеводов и карбоната кальция. (3) НУД основана на соотношении жира к углеводам и белку 0,6 : 1, содержит 60% жиров, 30% белка и 10% углеводов с низким гликемическим индексом (ГИ), ГИ < 50. ГИ измеряет тенденцию еды повышать уровень глюкозы в крови по сравнению с эквивалентным количеством эталонного углевода (глюкозы). По сравнению с классической КД, НУД дает меньшее повышение уровня кетоновых тел, но имеет сопоставимую эффективность, лучшую переносимость и более простую реализацию. · Рекомендуется применение кетогенной диеты детям с любой формой фармакорезистентной эпилепсии при неэффективности обычно применяемых противоэпилептических препаратов с целью альтернативного лечения эпилепсии [349, 535]. Уровень убедительности рекомендаций А (уровень достоверности доказательств – 1). Комментарии: Кетогенная терапия не должна быть терапией «отчаяния». Кетогенную диету следует рассмотреть как лечебную опцию у детей после неэффективности двух противоэпилептических препаратов, а при некоторых синдромах эпилепсии (при синдроме дефицита GLUT1 и пиридоксин-зависимой эпилепсии), как можно раньше. Показания [436] КД считается золотым стандартом для лечения метаболических заболеваний, таких как синдром дефицита белка-переносчика глюкозы 1 (GLUT-1) и дефицит пируватдегидрогеназы. В настоящее время КД показала высокую эффективность относительно лечения синдрома Веста. КД также может использовался при других эпилептических синдромах и является важным альтернативным лечением для пациентов с лекарственно-устойчивой эпилепсией, которые не являются кандидатами на хирургическое лечение. Эпилептические синдромы с вероятной эффективностью КД: синдром Ангельмана, митохондриопатии с нарушением комплекса 1, синдром Драве, МАЭ (синдром Доозе), синдром дефицита белка-переносчика глюкозы 1 (Glut-1), синдром эпилепсии, связанный с фебрильной инфекцией (FIRES), дети или младенцы на искусственном вскармливании (исключительно), инфантильные спазмы, синдром Отахара, дефицит пируватдегидрогеназы, суперрефрактерный эпилептический статус, туберозный склероз. Эпилептические синдромы с возможной эффективностью КД: дефицит аденилосукцинатлиазы, CDKL5-энцефалопатия, ДАЭ, пороки развития головного мозга, эпилепсия младенчества с мигрирующими фокальными приступами, эпилептическая энцефалопатия с продолженной спайк-волновой активностью во сне (CSWS), гликогеноз типа V,ЮМЭ, болезнь Лафора, синдром Ландау - Клеффнера, синдром Леннокса - Гасто, дефицит фосфофруктокиназы, синдром Ретта, подострый склерозирующий панэнцефалит. Абсолютные противопоказания Дефицит карнитина (первичный), дефицит карнитин-пальмитоилтрансферазы (CPT) I или II, дефицит карнитинтранслоказы, β-окислительные дефекты, дефицит среднецепочечной ацилдегидрогеназы (MCAD), дефицит длинноцепочечной ацилдегидрогеназы (LCAD), дефицит короткоцепочечной ацилдегидрогеназы (SCAD), дефицит длинноцепочечного 3-гидроксиацил-КоА, дефицит среднецепочечного 3-гидроксиацил-КоА, дефицит пируваткарбоксилазы, порфирия. К относительным противопоказаниям относятся неспособность поддерживать полноценное питание, возможное хирургическое лечение эпилепсии, низкая приверженность диеты родителями или опекуном, одновременное применение пропофола. Подготовка к введению КД Перед тем, как начать диету, пациент должен вести дневник приступов, чтобы установить параметр их частоты. Необходимо провести общий анализ крови с тромбоцитами, электролиты, КЩС включая бикарбонат сыворотки, общий белок, кальций, цинк, селен, магний и фосфаты, биохимический анализ крови (включая альбумин, аммиак и креатинин), липидный профиль натощак, ацилкарнитиновый профиль в сыворотке крови, общий анализ мочи, кальций и креатинин в моче, концентрацию противоэпилептического препарата, уровень витамина D. Определение органических кислот в моче и аминокислот в сыворотке крови рекомендуются в случае неясной природы эпилепсии. Также необходимы ЭЭГ и МРТ головного мозга. В случае риска нефролитиаза необходимо сделать УЗИ почек; электрокардиограмма и ультразвуковое исследование сонной артерии считаются необязательными. Оценка питания включает пищевой дневник, включая отчет о питании за 3 дня, пищевые привычки, аллергию, отвращение и непереносимость. Исходный вес, рост и идеальный вес для роста и индекса массы тела (ИМТ) необходимы для расчета кетогенного соотношения, калорий и потребления жидкости. Состав диеты следует подбирать в соответствии с возрастом пациента и способом кормления. Методика проведения КД Цель терапии состоит в том, чтобы достичь соотношения четырех частей жира к одной порции белка и углеводов, описываемого как «4 : 1». Чтобы достичь этого уровня, можно использовать один из двух подходов, с голоданием или без него. При первом подходе пациент должен быть госпитализирован на 12 – 48 часов или при наличии кетонов в моче, чтобы предотвратить развитие гипогликемии и обезвоживания. Этот метод имеет тенденцию ускорять развитие кетоза, хотя может вызвать больший стресс у пациента. Когда достигается кетоз, питание рассчитывается так, чтобы поддерживать постоянное соотношение КД. Последний подход не требует госпитализации, и коэффициент КД поэтапно увеличивается еженедельно с 1 : 1, 2 : 1 и 3 : 1 до 4 : 1. Большая часть литературы предполагает, что нет значительной разницы между двумя подходами с точки зрения времени, необходимого для достижения кетоза и возникновения гипогликемии, поэтому в настоящее время пациенты, как правило, не голодают перед началом КД. Принимая во внимание, что КД обеспечивает лишь небольшое количество фруктов, овощей, зерна, молока и сыра, необходимы пищевые добавки: мультивитаминные и минеральные комплексы с низким содержанием углеводов следует принимать ежедневно. К основным пищевым добавкам, необходимым для всех пациентов любого возраста, относят мультивитамины с минералами (включая микроэлементы, особенно селен), препараты кальция, витамин D и его аналоги (в соответствие с суточной потребностью, реже рекомендуют выше рекомендованной суточной нормы цитраты для перорального применения, слабительные средства для профилактики или лечения (лаурилсульфоацетат натрия, минеральное масло, глицериновые свечи), дополнительные селен, магний, цинк, фосфор, железо, медь, карнитин, масло MCT или кокосовое масло, соль (старше 1 года). Фонд Чарли (Charlie Foundation) разработал программу «KetoDietCalculator», чтобы помочь специалистам и лицам, осуществляющим уход, управлять диетической терапией. Эта онлайн-база данных рассчитывает планы питания для всех возрастов, от младенцев до взрослых, от твердой пищи до жидких смесей, и доступна в Интернете и на мобильных устройствах по ссылке http://www.ketodietcalculator.org. Наблюдение Пациентов, придерживающихся КД, следует регулярно осматривать каждые 3 месяца, и члены семьи или опекун должны иметь доступную связь с командой диетологов, чтобы разрешить возможные сомнения и обсудить побочные эффекты. При каждом визите следует оценивать дневник приступов, а также когнитивное развитие и поведение ребенка. При посещении клиники необходимы следующие обследования: оценка питания (дипломированный диетолог), роста, веса, идеальный вес для ИМТ, при необходимости - окружность головы у младенцев; оценка соответствия рецептов, используемых семьей или опекуном, рассчитанным калориям, белкам и жидкости; пересмотр дополнительных витаминных и минеральных добавок; оценка приверженности КД. При необходимости – изменение схемы рациона для улучшения приверженности диетическому лечению и контроля над приступами. Также необходимы: неврологический осмотр, оценка побочных эффектов КД, решение об изменении количества ПЭП (если применимо), оценка целесообразности дальнейшего продолжения КД. К лабораторной оценке относится общий анализ крови с тромбоцитами, электролиты, КЩС, включая бикарбонат сыворотки, общий белок, кальций, цинк, селен, магний и фосфаты, биохимический анализ крови (включая альбумин, аммиак и креатинин), липидный профиль натощак, ацилкарнитиновый профиль в сыворотке крови, общий анализ мочи, кальций и креатинин в моче, концентрацию ПЭП, уровень витамина D. ЭЭГ проводится для оценки эффективности КД или при рассмотрении вопроса о прекращении КД. При длительном нахождении на КД рекомендуется исследование минеральной плотности костной ткани (сканирование DEXA) через 2 года от начала КД. Следует помнить, что для достижения эффективности КД требуется период не менее 3 месяцев с момента достижения пациентом кетоза, поэтому важно побудить пациента и его семью продолжать диету в течение этого периода без изменения лекарства. Отмена КД Как указывалось ранее, КД следует использовать в среднем как минимум 3 месяца, чтобы сделать справедливую оценку эффективности терапии, прежде чем рассматривать вопрос о прекращении лечения. Если приступы учащаются через 1 - 2 недели после начала КД, диету следует немедленно прекратить. Если семья решает оставить своего ребенка на КД более 6 месяцев, несмотря на отсутствие очевидного контроля над приступами, решение в конечном итоге остается за ними и должно поддерживаться до тех пор, пока будут отслеживаться и устраняться побочные эффекты. У детей со снижением частоты приступов более чем на 50% КД прекращают ориентировочно через 2 года; однако у детей, у которых контроль приступов почти завершен (например, снижение приступов более чем на 90%) и побочные эффекты незначительны, КД можно продолжать в течение нескольких лет. Для КД нет максимальной продолжительности. Консенсусная группа рекомендует пересматривать риски и преимущества КД при каждом посещении клиники и, конечно, после 2 лет непрерывного использования. |
4. Медицинская реабилитация, медицинские показания и противопоказания к применению методов реабилитации
· Рекомендуется специалистам по реабилитации у больных эпилепсией старше 18 лет учитывать сниженную кардиореспираторную толерантность, с целью оценки модифицируемых факторов развития физических и психических хронических состояний и преждевременной смертности [437, 438].
Уровень убедительности рекомендаций С (уровень достоверности доказательств - 2).
· Рекомендуется проведение акупунктуры в сочетании с основной терапией для пациентов с эпилепсией для снижения частоты приступов [439].
Уровень убедительности рекомендаций А (уровень достоверности доказательств - 1).
· Рекомендуется проведение «школы эпилепсии» с обзором отдельных тем, связанных с эпилепсией, таких как типы приступы, управление стрессом, меры профилактики травм, эффективность лечения и побочные эффекты ПЭП членами междисциплинарной команды для пациентов с эпилепсией и их родственников для улучшения качества жизни пациентов с эпилепсией [440].
Уровень убедительности рекомендаций С (Уровень достоверности доказательств - 5).
· Рекомендуется организация наблюдения, включающего структурированное интерактивное обучающее приложение, организованное обученной медсестрой по эпилепсии, для пациентов с эпилепсией, для улучшения качества жизни, связанного со здоровьем, уменьшения дистресса, связанного с эпилепсией, беспокойства в связи с физическими и психическими эффектами ПЭП, а также в связи с физической активностью и работой [442, 443].
Уровень убедительности рекомендаций С (Уровень достоверности доказательств - 2).
· Рекомендуется использование навык-ориентированных психологических вмешательств (когнитивно-поведенческую терапию (КПТ), включая методы КПТ, такие как дыхательные приемы, рассуждение или визуализация; консультирование; и упражнения на осознанность) у взрослых и подростков с эпилепсией для улучшения качества жизни [443].
Уровень убедительности рекомендаций А. (Уровень достоверности доказательств - 2).
· Рекомендуется специалистам по реабилитации включить поведенческие методы в виде программ самоуправления в дополнении к стандартной терапии в комплексную помощь пациентам с эпилепсией с целью улучшении качества жизни, когнитивных функций и приверженности к лечению [444].
Уровень убедительности рекомендаций С (уровень достоверности доказательств - 5).
Комментарий: Когнитивно-повенденческая терапия представляется важным дополнением к стандартной противоэпилептической терапии при эпилепсии, особенно у пациентов с тревожно-депрессивными расстройствами [445]. Основной задачей когнитивно-поведенческой терапии при эпилепсии является коррекция иррациональных установок мышления пациентов, улучшение навыков совладания, самоэффективности и самоконтроля [446]. Программы когнитивно-поведенческой терапии включают обучение пациентов регулированию настроения, разрешению конфликтующих мыслей и эмоций («когнитивный диссонанс»), управлению стрессом и изменению образа жизни, чтобы минимизировать триггеры приступов [444]. Пациенты учатся определять неадаптивные модели мышления и заменять их более здоровыми когнитивными и поведенческими реакциями [447]. Обучение пациентов «эффективному самоуправлению» сводится к трем направлениям: осторожность изо дня в день, отрегулированный стиль жизни, информированное сознание [448].
· Рекомендуется рациональная психотерапия [449, 450].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 4).
Комментарий: Психотерапия при эпилепсии используется для влияния не только на патологические проявления, но и на личность пациента с целью приспособления к повседневной жизни в условиях изменившегося состояния здоровья. Психотерапевтические методы могут способствовать пониманию и смягчению аффективной напряженности, общему успокоению, формированию позитивного настроя у пациентов с эпилепсией. Психотерапия при эпилепсии позволяет более дифференцированно, гибко и «экономно» разрешать вопросы долгосрочного планирования медикаментозной терапии и реабилитации. Психотерапия при эпилепсии применяется в составе комплексного лечения при обязательном условии регулярной медикаментозной терапии [449]. Психотерапевтическая коррекция должна применяться адекватно клинической картине заболевания и с обязательным учетом личности пациента. Главными задачами рациональной психотерапии в виде беседы являются установление доверительного контакта с пациентом и разъяснительное подкрепление всех видов лечебных процедур и лекарственных назначений; изменение представления пациента о своей болезни; коррекция социальной установки, отношения к труду, окружающим. Во время психотерапевтической беседы необходимо обсуждать вопросы: как пациенты должны вести себя в обществе; подчеркивать необходимость участия их в трудовой деятельности; в виде советов рекомендовать, каким образом они могут учиться или работать [450].
4.2. Санаторно-курортное лечение, методы медицинской реабилитации, основанные на использовании природных лечебных факторов
В соответствии с приказом Министерства здравоохранения Российской Федерации от 28 сентября 2020 г. N 1029н «Об утверждении перечней медицинских показаний и противопоказаний для санаторно-курортного лечения» противопоказаниями к санаторно-курортному лечению являются [451]:
- Эпилепсия с текущими приступами, в том числе резистентная к проводимому лечению.
- Эпилепсия с ремиссией менее 6 месяцев (для санаторно-курортных организаций не психоневрологического профиля).
5. Профилактика и диспансерное наблюдение, медицинские показания и противопоказания к применению методов профилактики
По разным оценкам, до 25% случаев развития эпилепсии можно предотвратить путем первичной профилактики заболеваний и состояний, которые могут быть факторами риска эпилепсии. Предупреждение травм головы является самым эффективным способом предотвращения посттравматической эпилепсии. Надлежащая перинатальная помощь способствует уменьшению числа новых случаев эпилепсии, обусловленных перинатальной патологией ЦНС. Необходимы профилактиктические мероприятия, направленные на снижение заболеваемости и раннюю диагностику инфекционных и паразитарных заболеваний, являющихся факторами риска инфекционных эпилепсий.
· Рекомендуется генетическое консультирование и молекулярно-генетическое исследование мутации методом Сэнгера родителям пациента при выявлении у ребенка вероятно патогенной мутации или мутации с неясным клиническим значением в гене, ассоциированным с моногенным заболеванием, с целью прогноза деторождения [256].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5).
· Рекомендуется проведение профилактических мероприятий, направленных на снижение риска сердечно-сосудистых и цереброваскулярных заболеваний, а также диспансерное наблюдение за пациентами, перенесшими инсульт (контроль артериального давления, холестерина, глюкозы крови, снижение веса, а также отказ от курения и чрезмерного употребления алкоголя), для снижения риска постинсультной эпилепсии [452, 453].
Уровень убедительности рекомендаций С (уровень достоверности доказательств - 5).
· Рекомендуется диспансерное наблюдение пациентов с эпилепсией врачом-неврологом поликлиники или специализированного эпилептологического центра, проведение длительной противоэпилептической терапии с целью достижения ремиссии заболевания, сохранения оптимального качества жизни [6].
Уровень убедительности рекомендаций С (уровень достоверности доказательств — 5).
6. Организация оказания медицинской помощи
Медицинская помощь по профилю «эпилептология» осуществляется в виде:
- первичной медико-санитарной помощи;
- скорой медицинской помощи;
- специализированной медицинской помощи,
- высокотехнологичной медицинской помощи.
Медицинская помощь оказывается в следующих условиях:
- вне медицинской организации (по месту вызова бригады скорой медицинской помощи);
- амбулаторно (в условиях, не предусматривающих круглосуточное медицинское наблюдение и лечение);
- стационарно (в условиях, обеспечивающих круглосуточное медицинское наблюдение и лечение).
Первичная медико-санитарная помощь включает: первичную врачебную медико-санитарную помощь, первичную специализированную медико-санитарную помощь.
Первичная врачебная медико-санитарная помощь при эпилепсии в соответствии с Приказом Минздрава России от 24.12.2012 № 1404н [454] и Приказом Минздрава России от 24.12.2012 № 1439н [455] оказывается врачом-кардиологом, врачом-неврологом, врачом-психиатром, врачом-терапевтом, врачом-акушером-гинекологом, врачом-нейрохирургом, врачом-офтальмологом, врачом-эндокринологом, врачом-генетиком, врачом общей практики (семейным врачом), врачом-терапевтом участковым, врачом-эндокринологом в медицинских организациях.
Первичная специализированная медико-санитарная помощь при эпилепсии в соответствии с Приказом Минздрава России от 24.12.2012 № 1541н [456] оказывается врачом-акушером-гинекологом, врачом-генетиком, врачом-кардиологом, врачом-неврологом, врачом-нейрохирургом, врачом-офтальмологом, врачом-психиатром, врачом-терапевтом, врачом-эндокринологом в медицинских организациях, оказывающих специализированную, в том числе высокотехнологичную, медицинскую помощь.
Скорая медицинская помощь оказывается выездными бригадами скорой медицинской помощи в соответствии Приказом Минздрава России от 05.07.2016 N 468н [457]. Обязательной госпитализации подлежат пациенты с эпилептическим статусом, в состоянии комы и в состоянии с нарушением сознания в медицинские организации, оказывающие круглосуточную помощь и имеющие в своем составе отделения неотложной медицинской помощи.
Высокотехнологичная медицинская помощь осуществляется в соответствии с Приказом Министерства здравоохранения РФ от 10 декабря 2013 г. № 916н "О перечне видов высокотехнологичной медицинской помощи"[458].
При обращении пациента с эпилепсией к врачу общей практики, врачу-терапевту, кардиологу он направляется на консультацию к врачу-неврологу для определения плана обследования и назначения лечения. Большинство пациентов с эпилепсией могут проходить обследование и лечение у врача-невролога. Пациенты с эпилепсией состоят под наблюдением у врачей-неврологов.
В межрайонные (районные) специализированные кабинеты оказания помощи больным с эпилепсией и другими пароксизмальными состояниями и/или городские эпилептологические центры и/или областные эпилептологические центры (кабинеты) направляются больные для уточнения диагноза, назначения лечения и его коррекции.
Отбор больных для проведения консультаций в межрайонные (районные) специализированные кабинеты оказания помощи больным с эпилепсией и другими пароксизмальными состояниями, городские эпилептологические центры, областные эпилептологические центры (кабинеты) осуществляет врач-невролог или заведующий неврологического отделения медицинской организации.
Направлению в межрайонные (районные) специализированные кабинеты оказания помощи больным с эпилепсией и другими пароксизмальными состояниями и/или городские эпилептологические центры, областные эпилептологические центры (кабинеты) подлежат пациенты: с впервые выявленной эпилепсией; с ранее установленным диагнозом эпилепсии для коррекции лечения; при сохранении приступов на протяжении более 3 месяцев на фоне назначенной терапии [459]; с установленной резистентностью к проводимой терапии; с целью дифференциального диагноза эпилепсии с другими пароксизмальными состояниями; лица призывного возраста, имеющие в анамнезе пароксизмальные состояния; женщины с пароксизмальными состояниями, планирующие беременность.
При сохранении эпилептических приступов, развитии побочных эффектов, фармакорезистентных формах заболевания пациент может быть направлен в неврологический стационар; по показаниям в нейрохирургический стационар.
В неврологическом стационаре проводится поиск возможных причин «псевдорезистентности», проводится дифференциальный диагноз с псевдоэпилептическими приступами, пароксизмальными нарушениями сознания другой этиологии, с привлечением врачей других специальностей, уточняются типы эпилептических приступов, форма эпилепсии, осуществляется коррекция терапии, решается вопрос о возможности проведения «кетогенной диеты»,необходимости и возможности прехирургической подготовки и хирургического лечения пациентов с фармакорезистентным течением заболевания.
В стационаре должна быть возможность проведения нейрофизиологических методов обследования (ЭЭГ, видео-ЭЭГ-мониторинга), специализированного нейропсихологического тестирования, методов структурной нейровизуализации (МРТ-головного мозга, КТ головы), консультаций врачей специалистов (врача-психиатра, врача-кардиолога, врача-нейрохирурга и др). Только в условиях стационара возможно проведения видео-ЭЭГ-мониторинга со снижением доз либо отменой ПЭП с целью регистрации приступа (пароксизмального события) в случае проведения прехирургической подготовки пациентов с фармакорезистентными формами эпилепсии. При снижении доз ПЭП должна быть возможность оказания медицинской помощи в случае развития эпилептического статуса.
В нейрохирургический стационар направляются пациенты с фармакорезистентными формами эпилепсии для проведения прехирургической подготовки, в том числе с применением инвазивных электродных систем, и решения вопроса о нейрохирургическом лечении (резективных оперативных вмешательств, установке стимулятора блуждающего нерва и др.), проведении высокотехнологичной медицинской помощи.
Пациент с впервые выявленным эпилептическим приступом и пароксизмальным нарушением сознания подлежит обследованию и лечению в условиях стационара. С учётом конкретных клинико-анамнестических и данных рутинных лабораторно-инструментальных исследований врачу приемного отделения следует определить дальнейшую маршрутизацию пациента. При выявлении признаков эпилептического приступа, другой органической патологии центральной нервной системы показано продолжение обследования и лечения в условиях отделения неврологии. При выявлении признаков эпилептического статуса следует продолжить лечение в условиях отделения реанимации и интенсивной терапии.
Предположение психогенной природы пароксизмального события требует осмотра психиатра.
При веских доводах в пользу синкопального состояния пациент осматривается врачом-терапевтом (кардиологом) и направляется на лечение в терапевтическое (кардиологическое) отделение.
При наличии у пациента угрожающих нарушений сердечного ритма, гемодинамической нестабильности следует продолжить лечение в условиях отделения интенсивной терапии. По показаниям, осуществляется консультация врача-сердечно-сосудистого хирурга. При наличии обтурирующих интракардиальных образований, острой сердечно-сосудистой патологии пациента следует направить в отделение кардиохирургии или рентген-эндоваскулярных методов лечения.
7. Дополнительная информация (в том числе факторы, влияющие на исход заболевания или состояния)
Дополнительная информация отсутствует.
Критерии оценки качества медицинской помощи
Критерии оценки качества медицинской помощи
№
| Критерии качества | Выполнено/Не выполнено |
|---|---|---|
| Этап постановки диагноза | ДА/НЕТ |
1. | Определен (ы) для пациентов - взрослых и детей тип (ы) эпилептического приступа, форма эпилепсии и/или эпилептического синдрома, этиология эпилепсии и коморбидные состояния на основании классификации эпилептических приступов и эпилепсий МПЭЛ 2017 г. | ДА/НЕТ |
2. | Проведено неврологическое обследование для пациентов - взрослых и детей с впервые развившимся эпилептическим приступом | ДА/НЕТ |
3. | Проведена структурная МРТ головного мозга (не менее 1,5 тесла) пациентам – взрослым и детям, впервые в жизни перенесшим эпилептический приступ для выявления возможной причины заболевания ⃰ примечание: Исключение составляют пациенты с детской абсансной эпилепсией и возрастзависимой эпилепсией с центротемпоральными разрядами, если врач уверен в диагнозе. | ДА/НЕТ |
4. | Проведена по показаниям регистрация электрокардиограммы пациентам – взрослым и детям при подозрении на кардиогенную природу пароксизмальных расстройств. | ДА/НЕТ |
5. | Проведены лабораторные анализы пациентам – взрослым и детям с подозрением на эпилепсию по показаниям для проведения дифференциального диагноза и выявления декомпенсации соматических заболеваний: общий (клинический) анализ крови; анализ крови биохимический общетерапевтический (глюкоза, натрий, калий, кальций, магний, хлориды, аспартатаминотрансфераза, аланинаминотрансфераза и др.) | ДА/НЕТ |
6. | Проведена рутинная ЭЭГ с функциональными пробами пациентам - взрослым и детям с подозрением на эпилепсию | ДА/НЕТ |
7. | Проведена пациентам – взрослым и детям консультация врача-психиатра при подозрении на психогенную природу пароксизмальных состояний или на наличие у них коморбидных психических расстройств, поведенческих, эмоциональных, когнитивных нарушений* | ДА/НЕТ |
8. | Проведена консультация врача-кардиолога пациентам – взрослым и детям при подозрении на кардиогенную природу пароксизмальных расстройств*
| ДА/НЕТ |
| Этап лечения |
|
9. | Назначена противоэпилептическая терапия пациентам – взрослым и детям в режиме монотерапии в соответствии с формой эпилепсии и типом приступов, с учетом коморбидных заболеваний и принимаемых сопутствующих препаратов, при установлении диагноза «Эпилепсия» | ДА/НЕТ |
10. | Проведена коррекция противоэпилептической терапии пациентам – взрослым и детям: переход на режим альтернативной монотерапии или рациональной политерапии при сохранении приступов или появлении побочных эффектов у пациентов, принимающих первичную монотерапию ПЭП | ДА/НЕТ |
11. | Проведена МРТ головного мозга на аппарате 3 Т по эпилептологическому протоколу у пациентов – взрослых и детей с фармакорезистентной эпилепсией с целью уточнения этиологии заболевания и определения дальнейшей тактики лечения эпилепсии ⃰. | ДА/НЕТ |
12. | Проведена консультация врача-нейрохирурга пациентам с установленной фармакорезистентностью с целью решения вопроса о возможности нейрохирургического лечения эпилепсии ⃰. | ДА/НЕТ |
*Примечание: при невозможности осуществления рекомендации в данном учреждении пациент должен быть направлен в учреждение, где данная рекомендация может быть осуществлена.
При адекватном лечении возможно урежение или купирование эпилептических приступов и сохранение длительной ремиссии заболевания. Рекомендовано использовать термин «разрешение» эпилепсии при наличии следующих критериев:
1. Достижение определенного возраста у пациентов с зависящим от возраста эпилептическим синдромом;
2. Отсутствие эпилептических приступов в течение 10 лет у пациентов, не использовавших противоэпилептические препараты (ПЭП) не менее 5 лет.
В 1/3 случаях возможно формирование фармакорезистентности, прогрессирующее течение заболевания с учащением приступов и развитием когнитивных нарушений.
Эпилепсия представляет угрозу для жизни пациентов при наличии неконтролируемых приступов, отсутствии адекватной терапии и должного наблюдения за пациентом.
Список литературы
1. Fisher R.S. et al. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology // Epilepsia. Epilepsia, 2017. Vol. 58, № 4. P. 522–530.
2. Карлов В.А., Айвазян С.О. Эпилепсия в терминах, визуальных и ЭЭГ паттернах. Москва: АНО Учебный центр «Невромед-клиника», 2020. 72 p.
3. Fisher R.S. et al. ILAE Official Report: A practical clinical definition of epilepsy // Epilepsia. Blackwell Publishing Inc., 2014. Vol. 55, № 4. P. 475–482.
4. Trinka E. et al. A definition and classification of status epilepticus - Report of the ILAE Task Force on Classification of Status Epilepticus // Epilepsia. Blackwell Publishing Inc., 2015. Vol. 56, № 10. P. 1515–1523.
5. Scheffer I.E. et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology // Epilepsia. Wiley Online Library, 2017. Vol. 58, № 4. P. 512–521.
6. Карлов В.А. Эпилепсия у детей и взрослых женщин и мужчин. 2-е издание. ed. Москва: ООО "Издательский дом “БИНОМ,” 2019. 896 p.
7. Walker M.C. Hippocampal sclerosis: Causes and prevention // Semin. Neurol. Thieme Medical Publishers, Inc., 2015. Vol. 35, № 3. P. 193–200.
8. Malmgren K., Thom M. Hippocampal sclerosis-Origins and imaging // Epilepsia. John Wiley & Sons, Ltd, 2012. Vol. 53, № SUPPL. 4. P. 19–33.
9. Blümcke I. et al. International consensus classification of hippocampal sclerosis in temporal lobe epilepsy: A Task Force report from the ILAE Commission on Diagnostic Methods // Epilepsia. John Wiley & Sons, Ltd, 2013. Vol. 54, № 7. P. 1315–1329.
10. Blümcke I. et al. The clinicopathologic spectrum of focal cortical dysplasias: A consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission // Epilepsia. John Wiley & Sons, Ltd, 2011. Vol. 52, № 1. P. 158–174.
11. Najm I.M., Sarnat H.B., Blümcke I. Review: The international consensus classification of Focal Cortical Dysplasia – a critical update 2018 // Neuropathol. Appl. Neurobiol. Blackwell Publishing Ltd, 2018. Vol. 44, № 1. P. 18–31.
12. Slegers R.J., Blumcke I. Low-grade developmental and epilepsy associated brain tumors: A critical update 2020 // Acta Neuropathol. Commun. BioMed Central Ltd., 2020. Vol. 8, № 1.
13. Blümcke I. Epilepsy-associated brain tumors // Handbook of Clinical Neurology. Elsevier B.V., 2012. Vol. 108. P. 559–568.
14. Christensen J. The epidemiology of posttraumatic epilepsy // Semin. Neurol. Thieme Medical Publishers, Inc., 2015. Vol. 35, № 3. P. 218–222.
15. Lucke-Wold B.P. et al. Traumatic brain injury and epilepsy: Underlying mechanisms leading to seizure // Seizure. W.B. Saunders Ltd, 2015. Vol. 33. P. 13–23.
16. Zelano J. et al. How to diagnose and treat post-stroke seizures and epilepsy // Epileptic Disord. John Libbey, 2020. Vol. 22, № 3. P. 252–263.
17. Holtkamp M. et al. European Stroke Organisation guidelines for the management of post-stroke seizures and epilepsy // Eur. Stroke J. SAGE Publications Ltd, 2017. Vol. 2, № 2. P. 103–115.
18. Feyissa A.M., Hasan T.F., Meschia J.F. Stroke-related epilepsy // Eur. J. Neurol. Blackwell Publishing Ltd, 2019. Vol. 26, № 1. P. 18-e3.
19. Husari K.S., Dubey D. Autoimmune Epilepsy // Neurotherapeutics. Springer New York LLC, 2019. Vol. 16, № 3. P. 685–702.
20. Geis C. et al. Autoimmune seizures and epilepsy // J. Clin. Invest. American Society for Clinical Investigation, 2019. Vol. 129, № 3. P. 926–940.
21. Lemke J.R. et al. Targeted next generation sequencing as a diagnostic tool in epileptic disorders // Epilepsia. Epilepsia, 2012. Vol. 53, № 8. P. 1387–1398.
22. Рыжкова О.П. и др.. Руководство по интерпретации данных последовательности ДНК человека, полученных методами массового параллельного секвенирования (MPS) (редакция 2018, версия 2) // Медицинская генетика. Cifra Ltd - Russian Agency for Digital Standardization (RADS), 2019. Vol. 18, № 2. P. 3–23.
23. Pal D.K., Pong A.W., Chung W.K. Genetic evaluation and counseling for epilepsy // Nat. Rev. Neurol. Nat Rev Neurol, 2010. Vol. 6, № 8. P. 445–453.
24. Danzer S.C. Contributions of Adult-Generated Granule Cells to Hippocampal Pathology in Temporal Lobe Epilepsy: A Neuronal Bestiary // Brain Plast. / ed. Luikart B.W. 2018. Vol. 3, № 2. P. 169–181.
25. Forcelli P.A., Kalikhman D., Gale K. Delayed Effect of Craniotomy on Experimental Seizures in Rats // PLoS One. 2013. Т. 8. № 12. С. e81401
26. Gulyaeva N. V. Staging of neuroplasticity alterations during epileptogenesis (temporal lobe epilepsy as an example) // Zhurnal Nevrol. i psikhiatriiim. S.S. Korsakova. 2017. Т. 117. № 9. С. 10.
27. Klein P. идр. Commonalities in epileptogenic processes from different acute brain insults: Do they translate? // Epilepsia. 2018. Т. 59. № 1. С. 37–66.
28. Koyama R., Ikegaya Y. To BDNF or Not to BDNF: That Is the Epileptic Hippocampus // Neurosci. 2005. Т. 11. № 4. С. 282–287.
29. Liu Y.R. et al. Progressive Metabolic and Structural Cerebral Perturbations After Traumatic Brain Injury: An In Vivo Imaging Study in the Rat // J. Nucl. Med. 2010. Vol. 51, № 11. P. 1788–1795.
30. McNamara J.O., Scharfman H.E. Temporal lobe epilepsy and the BDNF receptor, TrkB // Epilepsia. 2010. Т. 51. № SUPPL. 5. С. 46–46.
31. O’Connor W.T., Smyth A., Gilchrist M.D. Animal models of traumatic brain injury: A critical evaluation // Pharmacol. Ther. 2011. Т. 130. № 2. С. 106–113.
32. Parent J.M., Kron M.M. Neurogenesis and epilepsy // Epilepsia. 2010. Vol. 51. P. 45–45.
33. Pitkänen A. et al. What Can We Model? // Models of Seizures and Epilepsy: Elsevier, 2017a. С. 1–3.
34. Pitkänen A. et al. Epilepsy After Traumatic Brain Injury // Models of Seizures and Epilepsy: Elsevier, 2017b. С. 661–681.
35. Raghupathi R. Cell Death Mechanisms Following Traumatic Brain Injury // Brain Pathol. 2004. Т. 14. № 2. С. 215–222.
36. Kharatishvili I. и др. A model of posttraumatic epilepsy induced by lateral fluid-percussion brain injury in rats // Neuroscience. 2006. Т. 140. № 2. С. 685–697.
37. Shorvon S.D. The etiologic classification of epilepsy // Epilepsia. 2011. Vol. 52, № 6. P. 1052–1057.
38. Thompson H.J. и др. Lateral Fluid Percussion Brain Injury: A 15-Year Review and Evaluation // J. Neurotrauma. 2005. Т. 22. № 1. С. 42–75.
39. Tran L.D. и др. Response of the Contralateral Hippocampus to Lateral Fluid Percussion Brain Injury // J. Neurotrauma. 2006. Т. 23. № 9. С. 1330–1342.
40. Vezzani A., Balosso S., Ravizza T. Inflammation and epilepsy // Handbook of Clinical Neurology. 2012. Vol. 107, № 1. P. 163–175.
41. WHO | Epilepsy: a public health imperative // WHO. World Health Organization, 2019.
42. Beghi E. et al. Global, regional, and national burden of epilepsy, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016 // Lancet Neurol. Lancet Publishing Group, 2019. Vol. 18, № 4. P. 357–375.
43. Commission on Epidemiology and Prognosis I.L.A.E. Guidelines for Epidemiologic Studies on Epilepsy // Epilepsia. 1993. Vol. 34, № 4. P. 592–596.
44. Fiest K.M. et al. Prevalence and incidence of epilepsy // Neurology. Lippincott Williams and Wilkins, 2017. Vol. 88, № 3. P. 296–303.
45. Guekht A. et al. The epidemiology of epilepsy in the Russian Federation // Epilepsy Res. Epilepsy Res, 2010. Vol. 92, № 2–3. P. 209–218.
46. Collaborators GBDE. Global, regional, and national burden of epilepsy, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. LancetNeurolog 2019;18(4):357–75.
47. Thurman D.J. et al. The burden of premature mortality of epilepsy in high-income countries: A systematic review from the Mortality Task Force of the International League Against Epilepsy // Epilepsia. Blackwell Publishing Inc., 2017. Vol. 58, № 1. P. 17–26.
48. Levira F. et al. Premature mortality of epilepsy in low- and middle-income countries: A systematic review from the Mortality Task Force of the International League Against Epilepsy // Epilepsia. Blackwell Publishing Inc., 2017. Vol. 58, № 1. P. 6–16.
49. Гусев Е.И. et al. Эпидемиология и медико-социальные аспекты болезней мозга в РФ. // Болезни мозга – медицинские и социальные аспекты – материалы международной конференции. / ed. Под редакцией Е.И. Гусева А.Б. Гехт. Москва: Буки-Веди, 2016. P. 27–46.
50. Зинчук М.С. et al. Суицидальность при эпилепсии: эпидемиологические аспекты и факторы риска // Журнал неврологии и психиатрии им. С.С. Корсакова. 2018. Vol. 118, № 2. P. 45–52.
51. Guekht A. Epilepsy, Comorbidities and Treatments // Curr. Pharm. Des. Bentham Science Publishers Ltd., 2017. Vol. 23, № 37. P. 5702–5726.
52. Holst A.G. et al. Epilepsy and risk of death and sudden unexpected death in the young: A nationwide study // Epilepsia. Epilepsia, 2013. Vol. 54, № 9. P. 1613–1620.
53. Leitinger M., Eugen Trinka E., Zimmermann G., Claudia A. Granbichler C.A., Kobulashvili T., Siebert U. Epidemiology of status epilepticus in adults: Apples, pears, and oranges — A critical review. M. Epilepsy&Behavior 103 (2020) 106720 doi.org/10.1016/j.yebeh.2019.106720 1525-5050
54. Shorvon S., Sen A. What is status epilepticus and what do we know about its epidemiology? // Seizure. Seizure, 2020. Vol. 75. P. 131–136.
55. Jette N. et al. ICD coding for epilepsy: Past, present, and future - A report by the International League Against Epilepsy Task Force on ICD codes in epilepsy // Epilepsia. Blackwell Publishing Inc., 2015. Vol. 56, № 3. P. 348–355.
56. Engel J. A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: Report of the ILAE task force on classification and terminology // Epilepsia. Epilepsia, 2001. Vol. 42, № 6. P. 796–803.
57. Fernandez-Baca Vaca G. et al. Epileptic seizure semiology in different age groups // Epileptic Disord. Epileptic Disorders, 2018. Vol. 20, № 3. P. 179–188.
58. Мухин К.Ю., Гатауллина С.Х., Петрухин А.С. Палеокортикальная височная эпилепсия, обусловленная мезиальным височным склерозом: клиника, диагностика и лечение (обзор литературы) // Русский журнал детской неврологии. Общество с ограниченной ответственностью «Издательский дом «АБВ-пресс», 2008. № 3. P. 41–60.
59. Takahashi A. et al. Frequent association of cortical dysplasia in dysembryoplastic neuroepithelial tumor treated by epilepsy surgery // Surg. Neurol. Surg Neurol, 2005. Vol. 64, № 5. P. 419–427.
60. Wagner J., Weber B., Elger C.E. Early and chronic gray matter volume changes in limbic encephalitis revealed by voxel-based morphometry // Epilepsia. Blackwell Publishing Inc., 2015. Vol. 56, № 5. P. 754–761.
61. Kotagal P. Treatment of Epilepsy: Principles and Practice / ed. Wylle E. Baltimore: Willams & Wilkins, 1997. 385–400 p.
62. King D. et al. Bilateral Hippocampal Atrophy in Medial Temporal Lobe Epilepsy // Epilepsia. Epilepsia, 1995. Vol. 36, № 9. P. 905–910.
63. Mohamed A. et al. Temporal lobe epilepsy due to hippocampal sclerosis in pediatric candidates for epilepsy surgery // Neurology. Neurology, 2001. Vol. 56, № 12. P. 1643–1649.
64. Chauvel P. Can we classify frontal lobe seizures? // Frontal lobe seizures and epilepsies in children. JOHN LIBBEY EUROTEXT, 2003. P. 59–64.
65. Мухин К.Ю. et al. Эпилептические синдромы. Диагностика и терапия. Руководство для врачей. Четвертое издание. ООО «Издательский дом «БИНОМ», 2018. 607p.
66. Roger J., Bureau M. Distinctive characteristics of frontral lobe epilepsy versus idiopathic generalized epilepsy // Adv. Neurol. 1992. Vol. 57. P. 399–410.
67. Мухин К.Ю., Петрухин А.С., Глухова Л.Ю. Эпилепсия. Атлас электро-клинической диагностики. Альварес Паблишинг, 2004. 364–388 p.
68. Salanova V., Andermann F. The Treatment of Epilepsy: Principles and Practice. Second edition. / ed. Wilye E. Willams & Wilkins, 1997. 423–431 p.
69. Holthausen H. et al. Structural (symptomatic) focal epilepsies of childhood. // Epileptic syndromes in infancy, childhood and adolescence. -5th edition with video. 5th ed. / ed. Bureau M., Delgado-Escueta A., Tassinari C. Paris, John Libbey Eurotext, 2012. P. 455–505.
70. Kahane P. et al. Perisylvian cortex involvement in seizures affecting the temporal lobe // Limbic seizures in children. John Libbey & Company Ltd, 2001. Vol. 8. P. 115–127.
71. Котов А.С. et al. Затылочная эпилепсия у взрослых // Журнал неврологии и психиатрии им. CC Корсакова. Общество с ограниченной ответственностью Издательство Медиа Сфера, 2009. Vol. 109, № 7. P. 4–8.
72. Usui N. et al. Posterior cortex epilepsy secondary to ulegyria: Is it a surgically remediable syndrome? // Epilepsia. Epilepsia, 2008. Vol. 49, № 12. P. 1998–2007.
73. Mukhin K.Y. Epileptic encephalopathies and related syndromes in children. John Libbey Eurotext, 2014. P. 429–448.
74. Walker M.C., Shorvon S.D. Status epilepticus and serial seizures // The treatment of epilepsy / ed. Shorvon S.D. et al. Oxford: Blackwell Science, 1995. P. 269–285.
75. Granata T. et al. Rasmussen’s encephalitis: Early characteristics allow diagnosis // Neurology. Neurology, 2003. Vol. 60, № 3. P. 422–425.
76. Hart Y., Andermann F. Rasmussen’s syndrome // Epileptic syndromes in infancy, childhood and adolescence / ed. Roger J. et al. John Libbey, 2005. P. 537–555.
77. Котов А.С. et al. Энцефалит Расмуссена. Описание двух клинических случаев // Русский журнал детской неврологии. Общество с ограниченной ответственностью «Издательский дом «АБВ-пресс», 2009. № 2. P. 42–51.
78. Bancaud J. Kojewnikows syndrome (epilepsia partialis continua) // Epileptic syndromes in infancy, childhood and adolescence. -2th edition with video. / ed. Bureau M. et al. London: John Libbey, 1992. P. 374–379.
79. Калинина Л.В. et al. Хронический прогрессирующий очаговый энцефалит Расмуссена // Журнал неврологии и психиатрии им. CC Корсакова. 1996. Vol. 2. P. 21–25.
80. Bien C.G., Elger C.E. Recent insights into Rasmussen encephalitis // Nervenarzt. Nervenarzt, 2005. Vol. 76, № 12. P. 1470–1487.
81. Hauser W.A. The Prevalence and Incidence of Convulsive Disorders in Children // Epilepsia. Epilepsia, 1994. Vol. 35. P. S1–S6.
82. Peiffer A. et al. A locus for febrile seizures (FEB3) maps to chromosome 2q23‐24 // Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. Wiley Online Library, 1999. Vol. 46, № 4. P. 671–678.
83. Scheffer I.E., Berkovic S.F. Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes // Brain. Oxford University Press, 1997. Vol. 120, № 3. P. 479–490.
84. Мухин К.Ю. etal. Фебрильные приступы (лекция) // Русский журнал детской неврологии. 2010. Vol. 5, № 2. P. 17–30.
85. Arzimanoglou A., Guerrini R., Aicardi J. Aicardi’s Epilepsy in children. 3rd ed. Philadelphia: Lippincott, 2004. 176–187 p.
86. Chauvel P., Dravet C. The HHE syndrome // Epileptic syndromes in infancy, childhood and adolescence. 4th edition. 4th ed. / ed. Roger J., Bureau M., Dravet C. John Libbey Eurotext, 2005. P. 277—293.
87. Elia M. et al. Myoclonic absence-like seizures and chromosome abnormality syndromes // Epilepsia. Epilepsia, 1998. Vol. 39, № 6. P. 660–663.
88. Tassinari C., Bureau M., Thomas P. Epilepsy with myoclonic absences // Epileptic syndromes in infancy, childhood and adolescence. London: John Libbey, 1992. P. 151–160.
89. Милованова О.А. et al. Эпилепсия с миоклоническими абсансами // Журнал неврологии и психиатрии им. CC Корсакова. Общество с ограниченной ответственностью Издательство Медиа Сфера, 1996. Vol. 96, № 2. P. 79–82.
90. Kelley S.A., Kossoff E.H. Doose syndrome (myoclonic-astatic epilepsy): 40 years of progress // Dev. Med. Child Neurol. Dev Med Child Neurol, 2010. Vol. 52, № 11. P. 988–993.
91. Мухин К.Ю., Миронов М.Б., Пылаева О.А. Эпилепсия с миоклонически-астатическими приступами (синдром Дозе) // Русский журнал детской неврологии. Общество с ограниченной ответственностью «Издательский дом «АБВ-пресс», 2013. № 1. P. 25–38.
92. Мухин К.Ю. Эпилепсия с миоклонически-астатическими приступами (синдром Доозе) // Идиопатические формы эпилепсии: систематика, диагностика, терапия. Арт-Бизнес- Центр, 2000. P. 150–157.
93. Doose H. Myoclonic astatic epilepsy of early childhood // Epileptic syndromes in infancy, childhood and adolescence. London: John Libbey, 1992. P. 103–114.
94. Мухин К.Ю. Детская абсанс эпилепсия // Идиопатические формы эпилепсии: систематика, диагностика, терапия / ed. Мухин К.Ю., Петрухин А.С. Арт-Бизнес-Центр, 2000. P. 63–72.
95. Loiseau T. Childhood absence epilepsy // Epileptic syndromes in infancy, childhood and adolescence. London: John Libbey, 1992. P. 135–150.
96. Obeid T., Awada A. Long-term prognosis of typical childhood absence epilepsy // Neurology. Neurology, 1997. Vol. 49, № 4. P. 1187.
97. Khan S., Al Baradie R. Epileptic encephalopathies: an overview // Epilepsy Research and Treatment. Epilepsy Research and Treatment, 2012. Vol. 2012.
98. Scheffer I.E., Liao J. Deciphering the concepts behind “Epileptic encephalopathy” and “Developmental and epileptic encephalopathy” // Eur. J. Paediatr. Neurol. W.B. Saunders Ltd, 2020. Vol. 24. P. 11–14.
99. Diagnostic Manual of International League Against Epilepsy [Electronic resource]. URL: https://www.epilepsydiagnosis.org/syndrome/epilepsy-syndrome-groupoverview.html.
100. Pressler R.M. et al. The ILAE classification of seizures and the epilepsies: Modification for seizures in the neonate. Position paper by the ILAE Task Force on Neonatal Seizures // Epilepsia. Epilepsia, 2021. Vol. 62, № 3. P. 615–628.
101. OMIM – Online. Mendelian Inheritance in Man [Electronic resource]. URL: https://www.ncbi.nlm.nih.gov/omim.
102. Morrison-Levy N. et al. Early-Onset Developmental and Epileptic Encephalopathies of Infancy: An Overview of the Genetic Basis and Clinical Features // Pediatr. Neurol. Elsevier Inc., 2021. Vol. 116. P. 85–94.
103. Мухин К.Ю. Юношеская абсансная эпилепсия // Идиопатические формы эпилепсии: систематика, диагностика, терапия / ed. Мухин К.Ю., Петрухин А.С. Арт-Бизнес-Центр, 2000. P. 75–80.
104. Obeid T. Clinical and genetic aspects of juvenile absence epilepsy // J. Neurol. Springer-Verlag, 1994. Vol. 241, № 8. P. 487–491.
105. Genton P., Gélisse P., Thomas P. Juvenile myoclonic epilepsy today: current definition and limits // Juvenile myoclonic epilepsy. The Janz syndrome. Wright Biomedical Publishing Ltd, 2000. P. 11–32.
106. Gilsoul M. et al. Subtle Brain Developmental Abnormalities in the Pathogenesis of Juvenile Myoclonic Epilepsy // Front. Cell. Neurosci. Frontiers Media S.A., 2019. Vol. 13. P. 1–10.
107. Panayiotopoulos C.P., Tahan R., Obeid T. Juvenile Myoclonic Epilepsy: Factors of Error Involved in the Diagnosis and Treatment // Epilepsia. Epilepsia, 1991. Vol. 32, № 5. P. 672–676.
108. Delgado-Escueta A. V., Enrile-Bacsal F. Juvenile myoclonic epilepsy of Janz // Neurology. Neurology, 1984. Vol. 34, № 3. P. 285–294.
109. Trenité D.G.A.K.-N. et al. Consensus on diagnosis and management of JME: from founder’s observations to current trends // Epilepsy Behav. Elsevier, 2013. Vol. 28. P. S87–S90.
110. Wolf P. et al. Juvenile myoclonic epilepsy: A system disorder of the brain // Epilepsy Res. Elsevier, 2015. Vol. 114. P. 2–12.
111. Janz D. The idiopathic generalized epilepsies of adolescence with childhood and juvenile age of onset // Epilepsia. Epilepsia, 1997. Vol. 38, № 1. P. 4–11.
112. Pellok J.M. The Differential Diagnosis of Epilepsy: Nonepileptic paroxysmal Disorders // The Treatment of Epilepsy: Principles and Practice / ed. Elaine W. Baltimore: Williams and Wilkins, 1996. P. 681 – 690.
113. Iivanainen M. Diagnosing epilepsy in patients with mental retardation // Epilepsy and mental retardation. / ed. Silapaa M. Biddles Ltd, 1999. P. 47–60.
114. Duncan J.S. Diagnosis – Is it epilepsy? // Clinical epilepsy / ed. Duncan J.S., Shorvon S.D., Fish D.R. Churchill Livingstone, 1995. P. 1–23.
115. Lowenstein D.H., Aminoff M.J., Simon R.P. Barbiturate anesthesia in the treatmentof status epilepticus: Clinical experience with 14 patients // Neurology. Neurology, 1988. Vol. 38, № 3. P. 395–400.
116. Холин А.А. et al. Эпилептический статус в младенческом и раннем детском возрасте // Автореферат дисс. доктора мед. наук. М. 2010.
117. Leitinger M. et al. Salzburg Consensus Criteria for Non-Convulsive Status Epilepticus - approach to clinical application // Epilepsy Behav. Epilepsy Behav, 2015. Vol. 49. P. 158–163.
118. Herman S.T. et al. Consensus statement on continuous EEG in critically Ill adults and children, part I: Indications // J. Clin. Neurophysiol. J Clin Neurophysiol, 2015. Vol. 32, № 2. P. 87–95.
119. Rubinos C., Reynolds A.S., Claassen J. The Ictal–Interictal Continuum: To Treat or Not to Treat (and How)? // Neurocrit. Care. Neurocrit Care, 2018. Vol. 29, № 1. P. 3–8.
120. Fiest K.M. et al. Depression in epilepsy: A systematic review and meta-analysis // Neurology. Neurology, 2013. Vol. 80, № 6. P. 590–599.
121. Siarava E. et al. Depression and quality of life in patients with epilepsy in Northwest Greece // Seizure. W.B. Saunders Ltd, 2019. Vol. 66. P. 93–98.
122. Josephson C.B. et al. Association of depression and treated depression with epilepsy and seizure outcomes a multicohort analysis // JAMA Neurol. American Medical Association, 2017. Vol. 74, № 5. P. 533–539.
123. Fazel S. et al. Premature mortality in epilepsy and the role of psychiatric comorbidity: A total population study // Lancet. Elsevier B.V., 2013. Vol. 382, № 9905. P. 1646–1654.
124. Mula M. et al. Psychiatric Comorbidities in People With Epilepsy // Neurol. Clin. Pract. Ovid Technologies (Wolters Kluwer Health), 2021. Vol. 11, № 2. P. 112–120.
125. Зинчук М.С. etal. Исследование суицидальности при эпилепсии: проблемы методологии // Журнал неврологии и психиатрии им. С.С. Корсакова. Спецвыпуски. Издательство ’МедиаСфера’, 2019. Vol. 119, № 11. P. 23–28.
126. Zinchuk M. et al. Validation of the Russian version of neurological disorders depression inventory for epilepsy (NDDI-E) // Epilepsy Behav. Academic Press Inc., 2020. Vol. 113, № 107549.
127. Kim D.H. et al. Optimal cutoff score of the Neurological Disorders Depression Inventory for Epilepsy (NDDI-E) for detecting major depressive disorder: A meta-analysis // Epilepsy and Behavior. Academic Press Inc., 2019. Vol. 92. P. 61–70.
128. Mula M. et al. Validation of rapid suicidality screening in epilepsy using the NDDIE // Epilepsia. Blackwell Publishing Inc., 2016. Vol. 57, № 6. P. 949–955.
129. Seo J.G. et al. Validation of the generalized anxiety disorder-7 in people with epilepsy: A MEPSY study // Epilepsy Behav. Academic Press Inc., 2014. Vol. 35. P. 59–63.
130. Micoulaud-Franchi J.A. et al. Rapid detection of generalized anxiety disorder and major depression in epilepsy: Validation of the GAD-7 as a complementary tool to the NDDI-E in a French sample // Epilepsy Behav. Academic Press Inc., 2016. Vol. 57, № Pt A. P. 211–216.
131. Gandy M. et al. Anxiety in epilepsy: A neglected disorder // J. Psychosom. Res. Elsevier Inc., 2015. Vol. 78, № 2. P. 149–155.
132. Wiglusz M.S., Landowski J., Cubała W.J. Validation of the Polish version of the Hospital Anxiety and Depression Scale for anxiety disorders in patients with epilepsy // Epilepsy Behav. Academic Press Inc., 2018. Vol. 84. P. 162–165.
133. Ross S. et al. Management of patients with newly diagnosed epilepsy: A systematic literature review // Am. Fam. Physician. Am Fam Physician, 2004. Vol. 70, № 5. P. 824,827-828.
134. King M.A. et al. Epileptology of the first-seizure presentation: A clinical, electroencephalographic, and magnetic resonance imaging study of 300 consecutive patients // Lancet. Elsevier B.V., 1998. Vol. 352, № 9133. P. 1007–1011.
135. Berg A.T. et al. Newly diagnosed epilepsy in children: Presentation at diagnosis // Epilepsia. Lippincott Williams and Wilkins, 1999. Vol. 40, № 4. P. 445–452.
136. Berg A. et al. Status epilepticus in children with newly diagnosed epilepsy // Ann. Neurol. 1999. Vol. 45. P. 618–623.
137. Sheldon R. et al. Historical criteria that distinguish syncope from seizures // J. Am. Coll. Cardiol. J Am Coll Cardiol, 2002. Vol. 40, № 1. P. 142–148.
138. Xu Y. et al. Frequency of a false positive diagnosis of epilepsy: A systematic review of observational studies // Seizure. W.B. Saunders Ltd, 2016. Vol. 41. P. 167–174.
139. Котов С. В. Эпилепсия у взрослых / С. В. Котов, И. Г. Рудакова, А. С. Котов. – Москва : "Пульс", 2008. – 332 с. – ISBN 9785934860579.
140. Scheepers B., Clough P., Pickles C. The misdiagnosis of epilepsy: Findings of a population study // Seizure. W.B. Saunders Ltd, 1998. Vol. 7, № 5. P. 403–406.
141. Smith D., Defalla B.A., Chadwick D.W. The misdiagnosis of epilepsy and the management of refractory epilepsy in a specialist clinic // QJM - Mon. J. Assoc. Physicians. Oxford University Press, 1999. Vol. 92, № 1. P. 15–23.
142. Hindley D., Ali A., Robson C. Diagnoses made in a secondary care “fits, faints, and funny turns” clinic // Arch. Dis. Child. Arch Dis Child, 2006. Vol. 91, № 3. P. 214–218.
143. Uldall P. et al. The misdiagnosis of epilepsy in children admitted to a tertiary epilepsy centre with paroxysmal events // Arch. Dis. Child. Arch Dis Child, 2006. Vol. 91, № 3. P. 219–221.
144. Wardrope A., Newberry E., Reuber M. Diagnostic criteria to aid the differential diagnosis of patients presenting with transient loss of consciousness: A systematic review // Seizure. W.B. Saunders Ltd, 2018. Vol. 61. P. 139–148.
145. Chen M. et al. Value of witness observations in the differential diagnosis of transient loss of consciousness // Neurology. Lippincott Williams and Wilkins, 2019. Vol. 92, № 9. P. E895–E904.
146. Doose H., Neubauer B.A. Preponderance of female sex in the transmission of seizure liability in idiopathic generalized epilepsy // Epilepsy Res. Epilepsy Res, 2001. Vol. 43, № 2. P. 103–114.
147. 1Avbersek A., Sisodiya S. Does the primary literature provide support for clinical signs used to distinguish psychogenic nonepileptic seizures from epileptic seizures? // Journal of Neurology, Neurosurgery and Psychiatry. J Neurol Neurosurg Psychiatry, 2010. Vol. 81, № 7. P. 719–725.
148. Dworetzky B.A. et al. Clinical characteristics of psychogenic nonepileptic seizure status in the long-term monitoring unit // Epilepsy Behav. Epilepsy Behav, 2006. Vol. 9, № 2. P. 335–338.
149. Azar N.J. et al. Postictal breathing pattern distinguishes epileptic from nonepileptic convulsive seizures // Epilepsia. Epilepsia, 2008. Vol. 49, № 1. P. 132–137.
150. Geyer J.D., Payne T.A., Drury I. The value of pelvic thrusting in the diagnosis of seizures and pseudoseizures // Neurology. Lippincott Williams and Wilkins, 2000. Vol. 54, № 1. P. 227–229.
151. Vinton A. et al. “Convulsive” nonepileptic seizures have a characteristic pattern of rhythmic artifact distinguishing them from convulsive epileptic seizures // Epilepsia. Epilepsia, 2004. Vol. 45, № 11. P. 1344–1350.
152. Chen D.K. et al. Sensitivity and specificity of video alone versus electroencephalography alone for the diagnosis of partial seizures // Epilepsy Behav. Epilepsy Behav, 2008. Vol. 13, № 1. P. 115–118.
153. Bell W.L. et al. Ictal cognitive assessment of partial seizures and pseudoseizures // Arch. Neurol. American Medical Association, 1998. Vol. 55, № 11. P. 1456–1459.
154. Slater J.D. et al. Induction of Pseudoseizures with Intravenous Saline Placebo // Epilepsia. Epilepsia, 1995. Vol. 36, № 6. P. 580–585.
155. Bianchi A. et al. Family study of epilepsy in first degree relatives: Data from the Italian Episcreen study // Seizure. W.B. Saunders Ltd, 2003. Vol. 12, № 4. P. 203–210.
156. Szaflarski J.P. et al. Seizure control in patients with epilepsy: The physician vs. medication factors // BMC Health Serv. Res. BMC Health Serv Res, 2008. Vol. 8.
157. Lowerison M.W. et al. Association of Levels of Specialized Care with Risk of Premature Mortality in Patients with Epilepsy // JAMA Neurol. American Medical Association, 2019. Vol. 76, № 11. P. 1352–1358.
158. Fisch L. et al. Early specialized care after a first unprovoked epileptic seizure // J. Neurol. Dr. Dietrich Steinkopff Verlag GmbH and Co. KG, 2016. Vol. 263, № 12. P. 2386–2394.
159. Ricci L. et al. Clinical utility of home videos for diagnosing epileptic seizures: a systematic review and practical recommendations for optimal and safe recording // Neurological Sciences. Springer-Verlag Italia s.r.l., 2021. Vol. 42, № 4. P. 1301–1309.
160. Tatum W.O. et al. Assessment of the Predictive Value of Outpatient Smartphone Videos for Diagnosis of Epileptic Seizures // JAMA Neurology. American Medical Association, 2020. Vol. 77, № 5. P. 593–600.
161. Beniczky S.A. et al. Seizure semiology inferred from clinical descriptions and from video recordings. How accurate are they? // Epilepsy Behav. Epilepsy Behav, 2012. Vol. 24, № 2. P. 213–215.
162. Ramanujam B., Dash D., Tripathi M. Can home videos made on smartphones complement video-EEG in diagnosing psychogenic nonepileptic seizures? // Seizure. W.B. Saunders Ltd, 2018. Vol. 62. P. 95–98.
163. Baykan B. et al. Does semiology tell us the origin of seizures consisting mainly of an alteration in consciousness? // Epilepsia. Epilepsia, 2011. Vol. 52, № 8. P. 1459–1466.
164. Beghi E. Management of a first seizure. General conclusions and recommendations // Epilepsia. Epilepsia, 2008. Vol. 49, № SUPPL. 1. P. 58–61.
165. Nowacki T.A., Jirsch J.D. Evaluation of the first seizure patient: Key points in the history and physical examination // Seizure. W.B. Saunders Ltd, 2017. Vol. 49. P. 54–63.
166. Camfield P.R. et al. Epilepsy after a first unprovoked seizure in childhood // Neurology. Neurology, 1985. Vol. 35, № 11. P. 1657–1660.
167. Wilson S.J. et al. Indications and expectations for neuropsychological assessment in routine epilepsy care: Report of the ILAE Neuropsychology Task Force, Diagnostic Methods Commission, 2013-2017 // Epilepsia. Blackwell Publishing Inc., 2015. Vol. 56, № 5. P. 674–681.
168. Velissaris S.L. et al. Psychological trajectories in the year after a newly diagnosed seizure // Epilepsia. Epilepsia, 2012. Vol. 53, № 10. P. 1774–1781.
169. Taylor J. et al. Patients with epilepsy: Cognitively compromised before the start of antiepileptic drug treatment? // Epilepsia. Epilepsia, 2010. Vol. 51, № 1. P. 48–56.
170. Witt J.A., Helmstaedter C. Should cognition be screened in new-onset epilepsies? A study in 247 untreated patients // J. Neurol. J Neurol, 2012. Vol. 259, № 8. P. 1727–1731.
171. Witt J.A. et al. Cognitive-behavioral screening in elderly patients with new-onset epilepsy before treatment // Acta Neurol. Scand. Blackwell Publishing Ltd, 2014. Vol. 130, № 3. P. 172–177.
172. Loughman A., Bowden S.C., D’Souza W. Cognitive functioning in idiopathic generalised epilepsies: A systematic review and meta-analysis // Neuroscience and Biobehavioral Reviews. Elsevier Ltd, 2014. Vol. 43. P. 20–34.
173. Rayner G., Wrench J.M., Wilson S.J. Differential contributions of objective memory and mood to subjective memory complaints in refractory focal epilepsy // Epilepsy Behav. Epilepsy Behav, 2010. Vol. 19, № 3. P. 359–364.
174. Oyegbile T.O. et al. The nature and course of neuropsychological morbidity in chronic temporal lobe epilepsy // Neurology. Lippincott Williams and Wilkins, 2004. Vol. 62, № 10. P. 1736–1742.
175. Baker G.A., Taylor J., Aldenkamp A.P. Newly diagnosed epilepsy: Cognitive outcome after 12 months // Epilepsia. Epilepsia, 2011. Vol. 52, № 6. P. 1084–1091.
176. Butler C.R., Zeman A.Z. Recent insights into the impairment of memory in epilepsy: Transient epileptic amnesia, accelerated long-term forgetting and remote memory impairment // Brain. Oxford University Press, 2008. Vol. 131, № 9. P. 2243–2263
177. Neligan A., Shorvon S.D. Prognostic factors, morbidity and mortality in tonic-clonic status epilepticus: A review // Epilepsy Research. Epilepsy Res, 2011. Vol. 93, № 1. P. 1–10.
178. Helmstaedter C. Cognitive outcome of status epilepticus in adults // Epilepsia. Epilepsia, 2007. Vol. 48, № SUPPL. 8. P. 85–90.
179. Tinuper P. et al. Epileptic drop attacks in partial epilepsy: Clinical features, evolution, and prognosis // J. Neurol. Neurosurg. Psychiatry. BMJ Publishing Group, 1998. Vol. 64, № 2. P. 231–237.
180. Parra-Díaz P., García-Casares N. Memory assessment in patients with temporal lobe epilepsy to predict memory impairment after surgery: A systematic review // Neurologia. Spanish Society of Neurology, 2019. Vol. 34, № 9. P. 596–606.
181. Harvey D.J. et al. Relationship between presurgical memory performance on the Wechsler Memory Scale-III and memory change following temporal resection for treatment of intractable epilepsy // Epilepsy Behav. Epilepsy Behav, 2008. Vol. 13, № 2. P. 372–375.
182. Äikiä M. et al. Verbal Memory in Newly Diagnosed Patients and Patients with Chronic Left Temporal Lobe Epilepsy // Epilepsy Behav. Epilepsy Behav, 2001. Vol. 2, № 1. P. 20–27.
183. LoGalbo A. et al. Verbal memory outcome in patients with normal preoperative verbal memory and left mesial temporal sclerosis // Epilepsy Behav. Epilepsy Behav, 2005. Vol. 6, № 3. P. 337–341.
184. Baxendale S., Thompson P.J., Sander J.W. Neuropsychological outcomes in epilepsy surgery patients with unilateral hippocampal sclerosis and good preoperative memory function // Epilepsia. Epilepsia, 2013. Vol. 54, № 9.
185. Sherman E.M.S. et al. Neuropsychological outcomes after epilepsy surgery: Systematic review and pooled estimates // Epilepsia. Epilepsia, 2011. Vol. 52, № 5. P. 857–869.
186. Helmstaedter C. Cognitive outcomes of different surgical approaches in temporal lobe epilepsy // Epileptic Disorders. Epileptic Disord, 2013. Vol. 15, № 3. P. 221–239.
187. Bjellvi J. et al. Complications of epilepsy surgery in Sweden 1996-2010: A prospective, population-based study // J. Neurosurg. American Association of Neurological Surgeons, 2015. Vol. 122, № 3. P. 519–525.
188. Van Schooneveld M.M.J., Braun K.P.J. Cognitive outcome after epilepsy surgery in children // Brain and Development. Brain Dev, 2013. Vol. 35, № 8. P. 721–729.
189. Helmstaedter C. et al. Disentangling the relationship between epilepsy and its behavioral comorbidities - The need for prospective studies in new-onset epilepsies // Epilepsy and Behavior. Epilepsy Behav, 2014. Vol. 31. P. 43–47.
190. Gilliam F., Kanner A.M. Treatment of depressive disorders in epilepsy patients // Epilepsy and Behavior. Academic Press Inc., 2002. Vol. 3, № 5 SUPPL. 1. P. 2–9.
191. Wiebe S, Téllez-Zenteno JF, Shapiro M. An evidence-based approach to the first seizure. Epilepsia. 2008; 49 Suppl 1:50-7. doi: 10.1111/j.1528-1167.2008.01451.x.
192. Hirtz D. et al. Practice parameter: Evaluating a first nonfebrile seizure in children: Report of the Quality Standards Subcommittee of the American Academy of Neurology, the Child Neurology Society, and the American Epilepsy Society // Neurology. Lippincott Williams and Wilkins, 2000. Vol. 55, № 5. P. 616–623.
193. Fletcher E.M., Sharieff G. Necessity of lumbar puncture in patients presenting with new onset complex febrile seizures // West. J. Emerg. Med. West J Emerg Med, 2013. Vol. 14, № 3. P. 206–211.
194. Sadek A.A. et al. Diagnostic value of lumbar puncture among infants and children presenting with fever and convulsions // Electron. physician. Mehr Publishing Group, 2016. Vol. 8, № 4. P. 2255–2262.
195. Vezzani A, Fujinami RS, White HS, Preux PM, Blümcke I, Sander JW, Löscher W. Infections, inflammation and epilepsy. Acta Neuropathol. 2016 Feb;131(2):211-234. doi: 10.1007/s00401-015-1481-5. Epub 2015 Sep 30. PMID: 26423537; PMCID: PMC4867498
196. Scramstad C., Jackson A.C. Cerebrospinal Fluid Pleocytosis in Critical Care Patients with Seizures // Can. J. Neurol. Sci. Cambridge University Press, 2017. Vol. 44, № 4. P. 343–349.
197. Blinder T., Lewerenz J. Cerebrospinal fluid findings in patients with autoimmune encephalitis-a systematic analysis // Front. Neurol. Frontiers Media S.A., 2019. Vol. 10, № JUL.
198. Cabezudo-García P. et al. Efficacy of antiepileptic drugs in autoimmune epilepsy: A systematic review // Seizure. W.B. Saunders Ltd, 2018. Vol. 59. P. 72–76.
199. Dubey D. et al. Neurological autoantibody prevalence in epilepsy of unknown etiology // JAMA Neurol. American Medical Association, 2017. Vol. 74, № 4. P. 397–402.
200. Dubey D. et al. Predictive models in the diagnosis and treatment of autoimmune epilepsy // Epilepsia. Blackwell Publishing Inc., 2017. Vol. 58, № 7. P. 1181–1189.
201. СарапуловаА.А., АйвазянС.О., ОсиповаК.В. Аутоиммунныеэнцефалопатии. Описание двух клинических случаев //QuantumSatis. – 2017. – Т. 1. – №. 2. – С. 72-83.
202. DelBruttoO.H. etal. Revised diagnostic criteria for neurocysticercosis // Journal of the Neurological Sciences. Elsevier B.V., 2017. Vol. 372. P. 202–210.
203. Reddy D.S., Volkmer R. Neurocysticercosis as an infectious acquired epilepsy worldwide // Seizure. W.B. Saunders Ltd, 2017. Vol. 52. P. 176–181.
204. Cattaneo D. et al. Association of HIV Infection with Epilepsy and Other Comorbid Conditions // AIDS Behav. Springer, 2020. Vol. 24, № 4. P. 1051–1055.
205. Лекции по ВИЧ-инфекции / под ред. В. В. Покровского. - 2-е изд., перераб. и доп. - М. : ГЭОТАР-Медиа, 2018. - 848 с. : ил. - ISBN 978- 5-9704-4374-3]
206. Sadeghi M. et al. An updated meta-analysis of the association between Toxoplasma gondii infection and risk of epilepsy // Trans. R. Soc. Trop. Med. Hyg. Oxford University Press, 2019. Vol. 113, № 8. P. 453–462.
207. Smithers-Sheedy H. et al. Congenital Cytomegalovirus among Children with Cerebral Palsy // J. Pediatr. Mosby Inc., 2017. Vol. 181. P. 267-271.e1.
208. Armangue T. et al. Frequency, symptoms, risk factors, and outcomes of autoimmune encephalitis after herpes simplex encephalitis: a prospective observational study and retrospective analysis // Lancet Neurol. Lancet Publishing Group, 2018. Vol. 17, № 9. P. 760–772.
209. Komaroff A.L., Pellett P.E., Jacobson S. Human herpesviruses 6a and 6b in brain diseases: Association versus causation // Clin. Microbiol. Rev. American Society for Microbiology, 2021. Vol. 34, № 1. P. 1–36.
210. ИерусалимскийА.П. Прогредиентныеформыклещевогоэнцефалита. — Новосибирск, 2011. — 76 с.
211. Taba P., Schmutzhard E., Forsberg P., Lutsar I., Ljøstad U., Mygland Å., Levchenko I., Strle F., Steiner I. EAN consensus review on prevention, diagnosis and management of tick-borne encephalitis. EurJNeurol. 2017; 24(10): 1214-e61
212. Haahr R. et al. Risk of neurological disorders in patients with european lyme neuroborreliosis: A nationwide, population-based cohort study // Clin. Infect. Dis. Oxford University Press, 2020. Vol. 71, № 6. P. 1511–1516.
213. Lee V.L.L. et al. Treatment, therapy and management of metabolic epilepsy: A systematic review // International Journal of Molecular Sciences. MDPI AG, 2018. Vol. 19, № 3.
214. Tomson T., Dahl M.L., Kimland E. Therapeutic monitoring of antiepileptic drugs for epilepsy // Cochrane Database of Systematic Reviews. John Wiley and Sons Ltd, 2007. № 2.
215. Patsalos P.N., Spencer E.P., Berry D.J. Therapeutic drug monitoring of antiepileptic drugs in epilepsy: A 2018 update // Therapeutic Drug Monitoring. Lippincott Williams and Wilkins, 2018. Vol. 40, № 5. P. 526–548.
216. Schoretsanitis G. et al. TDM in psychiatry and neurology: A comprehensive summary of the consensus guidelines for therapeutic drug monitoring in neuropsychopharmacology, update 2017; a tool for clinicians* // World Journal of Biological Psychiatry. Taylor and Francis Ltd, 2018. Vol. 19, № 3. P. 162–174.
217. Johannessen Landmark C., Johannessen S.I., Patsalos P.N. Therapeutic drug monitoring of antiepileptic drugs: current status and future prospects // Expert Opinion on Drug Metabolism and Toxicology. TaylorandFrancisLtd, 2020. Vol. 16, № 3. P. 227–238.
218. Якунина А.В., Повереннова И.Е. Роль лечебного мониторинга лекарственных средств при применении противоэпилептических препаратов. Эпилепсия и пароксизмальные состояния . 2016; 8 (3): 66-73. (В рус.) Https://doi.org/10.17749/2077-8333.2016.8.3.066-073].
219. Беляев О. В, Самыгин Д. Рекомендации экспертного совета по нейрофизиологии Российской противоэпилептической лиги по проведению рутинной ЭЭГ // Эпилепсия и пароксизмальные состояния. 2017. Vol. 8, № 4. P. 99–108.
220. Синкин М.В. et al. Русскоязычный словарь терминов, используемых в клинической электроэнцефалографии // Нервные болезни. Общество с ограниченной ответственностью «Издательское предприятие «Атмосфера», 2021. № 1. P. 83–88.
221. Hasan TF, Tatum WO. When should we obtain a routine EEG while managing people with epilepsy?. Epilepsy Behav Rep. 2021;16:100454. Published 2021 May 3. doi:10.1016/j.ebr.2021.100454
222. Glick T.H. The sleep-deprived electroencephalogram: Evidence and practice // Arch. Neurol. Arch Neurol, 2002. Vol. 59, № 8. P. 1235–1239.
223. King M.A. et al. Epileptology of the first-seizure presentation: A clinical, electroencephalographic, and magnetic resonance imaging study of 300 consecutive patients // Lancet. Elsevier B.V., 1998. Vol. 352, № 9133. P. 1007–1011.
224. MARSAN C.A., ZIVIN L.S. Factors Related to the Occurrence of Typical Paroxysmal Abnormalities in the EEG Records of Epileptic Patients MARSAN, C. A., & ZIVIN, L. S. (1970). Factors Related to the Occurrence of Typical Paroxysmal Abnormalities in the EEG Records of Epileptic Patie // Epilepsia. Epilepsia, 1970. Vol. 11, № 4. P. 361–381.
225. Baldin E. et al. Yield of epileptiform electroencephalogram abnormalities in incident unprovoked seizures: A population-based study // Epilepsia. Blackwell Publishing Inc., 2014. Vol. 55, № 9. P. 1389–1398.
226. Salinsky M., Kanter R., Dasheiff R.M. Effectiveness of Multiple EEGs in Supporting the Diagnosis of Epilepsy: An Operational Curve // Epilepsia. Epilepsia, 1987. Vol. 28, № 4. P. 331–334.
227. Koutroumanidis M. et al. The role of EEG in the diagnosis and classification of the epilepsy syndromes: a tool for clinical practice by the ILAE Neurophysiology Task Force (Part 1) // Epileptic Disord. Wiley Blackwell, 2017. Vol. 19, № 3. P. 233–298.
228. Burkholder D.B. et al. Routine vs extended outpatient EEG for the detection of interictal epileptiform discharges // Neurology. Lippincott Williams and Wilkins, 2016. Vol. 86, № 16. P. 1524–1530.
229. Loddenkemper T., Fernández I.S., Peters J.M. Continuous spike and waves during sleep and electrical status epilepticus in sleep // J. Clin. Neurophysiol. J Clin Neurophysiol, 2011. Vol. 28, № 2. P. 154–164.
230. ShethR D. Electroencephalogram in developmental delay: specific electroclinical syndromes SeminPediatr Neurol. 1998 Mar;5(1):45-51.
231. Beghi E. et al. Withdrawal of antiepileptic drugs: Guidelines of the Italian League Against Epilepsy // Epilepsia. Epilepsia, 2013. Vol. 54, № SUPPL.7. P. 2–12.
232. Rosenow F., Karl Martin Klein &Hajo M Hamer (2015) Non-invasive EEG evaluation in epilepsy diagnosis, Expert Review of Neurotherapeutics, 15:4, 425-444, DOI: 10.1586/14737175.2015.1025382
233. Baumgartner C. et al. Presurgical epilepsy evaluation and epilepsy surgery // F1000Research. F1000 Research Ltd, 2019. Vol. 8.
234. West S., Nolan S.J., Newton R. Surgery for epilepsy: A systematic review of current evidence // Epileptic Disord. John Libbey Eurotext, 2016. Vol. 18, № 2. P. 113–121.
235. Seeck M. et al. The standardized EEG electrode array of the IFCN // Clin. Neurophysiol. Elsevier Ireland Ltd, 2017. Vol. 128, № 10. P. 2070–2077.
236. Usman S.M. et al. Using scalp EEG and intracranial EEG signals for predicting epileptic seizures: Review of available methodologies // Seizure. W.B. Saunders Ltd, 2019. Vol. 71. P. 258–269.
237. Климчук О.В. et al. Стандарты выполнения магнитно-резонансной томографии головного мозга для диагностики эпилепсии. Государственное бюджетное учреждение здравоохранения города Москвы" Научно …, 2016. P. 15.
238. Авакян Г.Н. et al. Рекомендации Российской Противоэпилептической Лиги (РПЭЛ) по использованию магнитно-резонансной томографии в диагностике эпилепсии // Эпилепсия и пароксизмальные состояния. Общество с ограниченной ответственностью «Ирбис», 2019. Vol. 11, № 3. P. 208–232.
239. Bernasconi A. et al. Recommendations for the use of structural magnetic resonance imaging in the care of patients with epilepsy: A consensus report from the International League Against Epilepsy Neuroimaging Task Force // Epilepsia. Blackwell Publishing Inc., 2019. Vol. 60, № 6. P. 1054–1068.
240. Duncan J.S. et al. Brain imaging in the assessment for epilepsy surgery // Lancet Neurol. Lancet Publishing Group, 2016. Vol. 15, № 4. P. 420–433.
241. Craven I. et al. 3.0 T MRI of 2000 consecutive patients with localisation-related epilepsy // Br. J. Radiol. BrJRadiol, 2012. Vol. 85, № 1017. P. 1236–1242.
242. Крылов В.В. etal. Хирургическое лечение больных с магнитно-резонансно-негативными фармакорезистентными формами эпилепсии // Неврологический журнал. ОАО «Издательство «Медицина», 2016. Vol. 21, № 4. P. 213–218.
243. Крылов В.В. et al. Клинические рекомендации по предоперационному обследованию и хирургическому лечению пациентов с фармакорезистентными формами эпилепсии // Неврологический журнал. 2015. Vol. 21, № 4. P. 213–218.
244. Крылов В.В. et al. Результаты хирургического лечения пациентов с фармакорезистентными формами эпилепсии // Нейрохирургия. 2017. № 1. P. 15–22.
245. Nakae S. et al. Association of preoperative seizures with tumor metabolites quantified by magnetic resonance spectroscopy in gliomas // Sci. Rep. Nature Research, 2021. Vol. 11, № 1.
246. Шалькевич Л.В., Кудлач А.И. Электроэнцефалографические и нейровизуализационные предикторы развития эпилепсии после однократного неспровоцированного приступа у детей // Неврология и нейрохирургия. Восточная Европа. Издательское частное унитарное предприятие" Профессиональные издания", 2016. Vol. 6, № 2. P. 189–196.
248. Abdelgawad E.A. et al. Magnetic resonance imaging (MRI) volumetry in children with nonlesional epilepsy, does it help? // Egypt. J. Radiol. Nucl. Med. Springer Open, 2021. Vol. 52, № 35. P. 1–11.
249. Bernasconi N., Wang I. Emerging Trends in Neuroimaging of Epilepsy // Epilepsy Curr. SAGE Publications Ltd, 2021. Vol. 21, № 2. P. 79–82.
250. Tranvinh E. et al. Imaging evaluation of the adult presenting with new-onset seizure // Am. J. Roentgenol. AmericanRoentgenRaySociety, 2019. Vol. 212, № 1. P. 15–25.
251. Евстигнеев В.В., Сакович Р.А., Кистень О.В. Результаты первого опыта использования позитронно-эмиссионной томографии в Республике Беларусь для диагностики эпилепсии // Международный неврологический журнал. ФЛП «Заславский Александр Юрьевич», 2017. Vol. 3, № 89. P. 110–116.
252. Tang Y. Et al. FDG-PET Profiles of Extratemporal Metabolism as a Predictor of Surgical Failure in Temporal Lobe Epilepsy // Front. Med. Frontiers Media S.A., 2020. Vol. 7.
253. Chassoux F. et al. 18F-FDG-PET patterns of surgical success and failure in mesial temporal lobe epilepsy // Neurology. Lippincott Williams and Wilkins, 2017. Vol. 88, № 11. P. 1045–1053.
254. Stefanski A. et al. Clinical sequencing yield in epilepsy, autism spectrum disorder, and intellectual disability: A systematic review and meta-analysis // Epilepsia. Blackwell Publishing Inc., 2021. Vol. 62, № 1. P. 143–151.
255. Аношкин К.И., Карандашева К.О., Танас А.С., Бесонова Л.А., Демина Н.А., Петухова М.С., Анисимова И.В., Залетаев Д.В., Стрельников В.В. Медицинская технология комплексной ДНК-диагностики туберозного склероза. Медицинская генетика, 2018, Том 17 №8, стр. 32-37
256. Ritter D.M., Holland K. Genetic Testing in Epilepsy // Semin. Neurol. Thieme Medical Publishers, Inc., 2020. Vol. 40, № 6. P. 730–738.
257. Anney R.J.L. et al. Genetic determinants of common epilepsies: A meta-analysis of genome-wide association studies // Lancet Neurol. Lancet Publishing Group, 2014. Vol. 13, № 9. P. 893–903.
258. Singh R. et al. Chromosomal abnormalities and epilepsy: A review for clinicians and gene hunters // Epilepsia. Epilepsia, 2002. Vol. 43, № 2. P. 127–140.
259. Coppola A. et al. Diagnostic implications of genetic copy number variation in epilepsy plus // Epilepsia. Blackwell Publishing Inc., 2019. Vol. 60, № 4. P. 689–706.
260. Berkovic S.F. et al. The Epilepsy Genetics Initiative: Systematic reanalysis of diagnostic exomes increases yield // Epilepsia. Blackwell Publishing Inc., 2019. Vol. 60, № 5. P. 797–806.
261. Bonello D., Camilleri F., Calleja-AgiusJ. Angelman Syndrome: Identification and Management Neonatal Netw. 2017 May 1;36(3):142-151.
262. Байдакова Г.В., Иванова Т.А., Захарова Е.Ю., Кокорина О.С. Роль тандемной масс-спектрометрии в диагностике наследственных болезней обмена веществ // РЖДГиО. 2018. №3.
263. Campistol J. Epilepsy in Inborn Errors of Metabolism With Therapeutic Options // Semin. Pediatr. Neurol. W.B. Saunders, 2016. Vol. 23, № 4. P. 321–331.
264. van Karnebeek CD, Stockler S. Treatable inborn errors of metabolism causing intellectual disability: a systematic literature review. Mol Genet Metab. 2012 Mar;105(3):368-81. doi: 10.1016/j.ymgme.2011.11.191. Epub 2011 Nov 30
265. Wilmshurst J.M. et al. Summary of recommendations for the management of infantile seizures: Task Force Report for the ILAE Commission of Pediatrics // Epilepsia. Blackwell Publishing Inc., 2015. Vol. 56, № 8. P. 1185–1197.
266. Gillis L., Kaye E. Diagnosis and management of mitochondrial diseases // Pediatric Clinics of North America. Pediatr Clin North Am, 2002. Vol. 49, № 1. P. 203–219.
267. Mulley GC, Mefford H.C. Epilepsy and the new cytogenetics. Epilepsia2011 Mar;52(3):423-32.
268. Fraser L.K. et al. Rising national prevalence of life-limiting conditions in children in England // Pediatrics. Pediatrics, 2012. Vol. 129, № 4.
269. Hain R. et al. Paediatric palliative care: Development and pilot study of a “Directory” of life-limiting conditions // BMC Palliat. Care. BMCPalliatCare, 2013. Vol. 12, № 1.
270. Вишняков Н.И. etal. Медико-социальные проблемы хосписов в педиатрической практике Санкт-Петербурга // Педиатр. Общество с ограниченной ответственностью «Эко-Вектор», 2014. Vol. 5, № 1.
271. Dallas J., Englot D.J., Naftel R.P. Neurosurgical approaches to pediatric epilepsy: Indications, techniques, and outcomes of common surgical procedures // Seizure. W.B. Saunders Ltd, 2020. Vol. 77. P. 76–85.
272. Лебедев К.Э., Маматханов М.Р. Показания и общие принципы хирургического лечения эпилепсии (обзор) // Нейрохирургия и неврология детского возраста. Федеральное государственное учреждение" Российский научно-исследовательский …, 2016. № 2. P. 66–78.
273. Макнамара-Гуджер К. и Фойдтнер К. Глава 1. История и эпидемиология. С. 5. Паллиативная помощь детям. Под редакцией Э. Голдман, Р. Хейна и С. Либена. Пер с англ. – М.: Практика, 2017. – 672 с.
274. Mattson R.H. et al. Comparison of Carbamazepine, Phenobarbital, Phenytoin, and Primidone in Partial and Secondarily Generalized Tonic–Clonic Seizures // N. Engl. J. Med. New England Journal of Medicine (NEJM/MMS), 1985. Vol. 313, № 3. P. 145–151.
275. Nevitt S.J. et al. Antiepileptic drug monotherapy for epilepsy: a network meta-analysis of individual participant data // Cochrane Database Syst. Rev. Wiley, 2017. Vol. 12, № 12.
276. Lattanzi S. et al. Antiepileptic monotherapy in newly diagnosed focal epilepsy. A network meta-analysis // Acta Neurologica Scandinavica. Blackwell Publishing Ltd, 2019. Vol. 139, № 1. P. 33–41.
277. Campos M.S. de A. et al. Efficacy and Tolerability of Antiepileptic Drugs in Patients with Focal Epilepsy: Systematic Review and Network Meta-analyses // Pharmacotherapy. Pharmacotherapy Publications Inc., 2016. Vol. 36, № 12. P. 1255–1271.
278. Baulac M. et al. Efficacy, safety, and tolerability of lacosamide monotherapy versus controlled-release carbamazepine in patients with newly diagnosed epilepsy: a phase 3, randomised, double-blind, non-inferiority trial // Lancet Neurol. Lancet Publishing Group, 2017. Vol. 16, № 1. P. 43-54.
279. Marson A.G. et al. Carbamazepine versus valproate monotherapy for epilepsy // Cochrane Database Syst. Rev. Wiley, 2000. Vol. 2000, № 3.
280. Nevitt S.J. et al. Topiramate versus carbamazepine monotherapy for epilepsy: An individual participant data review // Cochrane Database of Systematic Reviews. John Wiley and Sons Ltd, 2019. Vol. 2019, № 6.
281. Nevitt S.J. et al. Lamotrigine versus carbamazepine monotherapy for epilepsy: An individual participant data review // Cochrane Database of Systematic Reviews. John Wiley and Sons Ltd, 2018. Vol. 2018, № 6.
282. Brodie M.J. et al. Comparison of levetiracetam and controlled-release carbamazepine in newly diagnosed epilepsy // Neurology. Neurology, 2007. Vol. 68, № 6. P. 402–408.
283. Trinka E. et al. KOMET: An unblinded, randomised, two parallelgroup, stratified trial comparing the effectiveness of levetiracetam with controlled-release carbamazepine and extended-release sodium valproate as monotherapy in patients with newly diagnosed epilepsy // J. Neurol. Neurosurg. Psychiatry. BMJ Publishing Group, 2013. Vol. 84, № 10. P. 1138–1147.
284. Marson A. et al. The SANAD study of effectiveness of valproate, lamotrigine, or topiramate for generalised and unclassifiable epilepsy: An unblinded randomised controlled trial // Lancet. 2007. Vol. 369. P. 1016–1026.
285. Glauser T. et al. Updated ILAE evidence review of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes // Epilepsia. Epilepsia, 2013. Vol. 54, № 3. P. 551–563.
286. Brigo F. et al. Clonazepam monotherapy for treating people with newly diagnosed epilepsy // Cochrane Database of Systematic Reviews. John Wiley and Sons Ltd, 2019. Vol. 2019, № 11.
287. Lattanzi S. et al. Antiepileptic drug monotherapy for epilepsy in the elderly: A systematic review and network meta-analysis // Epilepsia. Blackwell Publishing Inc., 2019. Vol. 60, № 11. P. 2245–2254.
288. Rowan A.J. et al. New onset geriatric epilepsy: A randomized study of gabapentin, lamotrigine, and carbamazepine // Neurology. Lippincott Williams and Wilkins, 2005. Vol. 64, № 11. P. 1868–1873.
289. Авакян Г.Н. et al. Ограничения использования вальпроевой кислоты у девочек и женщин: расширение противопоказаний в инструкции по медицинскому применению, основанное на данных реальной клинической практики // Эпилепсия и пароксизмальные состояния. Общество с ограниченной ответственностью «Ирбис», 2019. Vol. 11, № 2.
290. Liu J., Wang L.N., Wang Y.P. Topiramatemonotherapyforjuvenilemyoclonicepilepsy // CochraneDatabaseofSystematicReviews. John Wiley and Sons Ltd, 2017. Vol. 2017, № 4.
291. Wiebe S., Jette N. Pharmacoresistance and the role of surgery in difficult to treat epilepsy // Nat. Rev. Neurol. Nat Rev Neurol, 2012. Vol. 8, № 12. P. 669–677.
292. Bodalia P.N. et al. Comparative efficacy and tolerability of anti-epileptic drugs for refractory focal epilepsy: Systematic review and network meta-analysis reveals the need for long term comparator trials // Br. J. Clin. Pharmacol. Br J Clin Pharmacol, 2013. Vol. 76, № 5. P. 649–667.
293. Ma J., Huang S., You C. Adjunctive brivaracetam for patients with refractory partial seizures: A meta-analysis of randomized placebo-controlled trials // Epilepsy Res. Elsevier, 2015. Vol. 114. P. 59–65.
294. French J.A. et al. Evaluation of adjunctive perampanel in patients with refractory partial-onset seizures: Results of randomized global phase III study 305 // Epilepsia. Epilepsia, 2013. Vol. 54, № 1. P. 117–125.
295. Elger C. et al. Efficacy and safety of eslicarbazepine acetate as adjunctive treatment in adults with refractory partial-onset seizures: A randomized, double-blind, placebo-controlled, parallel-group phase III study // Epilepsia. Epilepsia, 2009. Vol. 50, № 3. P. 454–463.
296. Song L. et al. Clonazepam add-on therapy for drug-resistant epilepsy // Cochrane Database of Systematic Reviews. John Wiley and Sons Ltd, 2020. Vol. 2020, № 4.
297. Colleran N., O Connor T., O Brien J.J. Anti epileptic drug trials for patients with drug resistant idiopathic generalised epilepsy: A meta-analysis // Seizure. W.B. Saunders Ltd, 2017. Vol. 51. P. 145–156.
298. Marson A. et al. The SANAD II study of the effectiveness and cost-effectiveness of valproate versus levetiracetam for newly diagnosed generalised and unclassifiable epilepsy: an open-label, non-inferiority, multicentre, phase 4, randomised controlled trial // Lancet. Elsevier B.V., 2021. Vol. 397, № 10282. P. 1375–1386.
299. Воронкова К.В. и др. Современный выбор антиэпилептической терапии: этапы и рекомендации //Эпилепсия и пароксизмальные состояния. – 2018. – Т. 10. – №. 2.
300. Booth D., Evans D.J. Anticonvulsants for neonates with seizures // Cochrane Database Syst. Rev. Wiley, 2004. № 4.
301. Ijff D.M., Aldenkamp A.P. Cognitive side-effects of antiepileptic drugs in children // Handbook of Clinical Neurology. Elsevier B.V., 2013. Vol. 111. P. 707–718.
302. Bensch J. et al. A Double‐blind Study of Clonazepam in the Treatment of Therapy‐resistant Epilepsy in Children // Dev. Med. Child Neurol. Dev Med Child Neurol, 1977. Vol. 19, № 3. P. 335–342.
303. Guerreiro M.M. et al. A double-blind controlled clinical trial of oxcarbazepine versus phenytoin in children and adolescents with epilepsy // Epilepsy Res. Epilepsy Res, 1997. Vol. 27, № 3. P. 205–213.
304. Glauser TA et al. Adjunctive therapy with oxcarbazepine in children with partial seizures. The Oxcarbazepine Pediatric Study Group. Neurology. 2000; 54(12):2237–44.
305. Bittencourt PR, Antoniuk SA, Bigarella MM, da Costa JC, Doro MP, Ferreira AS, et al. Carbamazepine and phenytoin in epilepsies refractory to barbiturates: efficacy, toxicity and mental function. Epilepsy Res. 1993;16(2):147–55
306. Wheless J.W. et al. Treatment of pediatric epilepsy: European expert opinion, 2007 // Epileptic Disord. Epileptic Disord, 2007. Vol. 9, № 4. P. 353–412.
307. Pina-Garza J.E.; Nordli D.R.; Rating D.; Yang H.; Schiemann-Delgado J.; Duncan B.; Adjunctive levetiracetam in infants and young children with refractory partial-onset seizures; Epilepsia;2009;50;5:1141-1149
308. Glauser T.A. et al. Double-blind placebo-controlled trial of adjunctive levetiracetam in pediatric partial seizures // Neurology. Neurology, 2006. Vol. 66, № 11. P. 1654–1660.
309. Rosenfeld W.E. et al. Levetiracetam as add-on therapy for idiopathic generalized epilepsy syndromes with onset during adolescence: Analysis of two randomized, double-blind, placebo-controlled studies // Epilepsy Res. Epilepsy Res, 2009. Vol. 85, № 1. P. 72–80.
310. Arzimanoglou A, Ferreira J, Satlin A, Olhaye O, Kumar D, Dhadda S, Bibbiani F. Evaluation of long-term safety, tolerability, and behavioral outcomes with adjunctive rufinamide in pediatric patients (≥1 to <4 years old) with Lennox-Gastaut syndrome: Final results from randomized study 303. Eur J Paediatr Neurol. 2019 Jan;23(1):126-135.
311. Ohtsuka Y. et al. Rufinamide as an adjunctive therapy for Lennox-Gastaut syndrome: A randomized double-blind placebo-controlled trial in Japan // Epilepsy Res. Elsevier B.V., 2014. Vol. 108, № 9. P. 1627–1636.
312. Gilliam FG, Veloso F, Bomhof MA, Gazda SK, Biton V, Ter Bruggen JP, Neto W, Bailey C, Pledger G, Wu SC. A dose-comparison trial of topiramate as monotherapy in recently diagnosed partial epilepsy. Neurology. 2003 Jan 28;60(2):196-202. doi: 10.1212/01.wnl.0000048200.12663.bc. PMID: 12552030.
313. Sachdeo R.C. et al. A double-blind, randomized trial of topiramate in Lennox-Gastaut syndrome // Neurology. Lippincott Williams and Wilkins, 1999. Vol. 52, № 9. P. 1882–1887.
314. Frank L.M. et al. Lamictal (lamotrigine) monotherapy for typical absence seizures in children // Epilepsia. Lippincott Williams and Wilkins, 1999. Vol. 40, № 7. P. 973–979.
315. Smith D, Baker G, Davies G, Dewey M, Chadwick DW. Outcomes of add-on treatment with lamotrigine in partial epilepsy. Epilepsia. 1993 Mar-Apr;34(2):312-22. doi: 10.1111/j.1528-1157.1993.tb02417.x. PMID: 8453943
316. Duchowny M. et al. A placebo-controlled trial of lamotrigine add-on therapy for partial seizures in children // Neurology. Lippincott Williams and Wilkins, 1999. Vol. 53, № 8. P. 1724–1731.
317. Trevathan E. et al. Lamotrigine adjunctive therapy among children and adolescents with primary generalized tonic-clonic seizures // Pediatrics. Pediatrics, 2006. Vol. 118, № 2.
318. Motte J. et al. Lamotrigine for Generalized Seizures Associated with the Lennox–Gastaut Syndrome // N. Engl. J. Med. New England Journal of Medicine (NEJM/MMS), 1997. Vol. 337, № 25. P. 1807–1812.
319. Белоусова Е.Д., Шулякова И.В., Охапкина Т.Г. Гормональная терапия синдрома Веста // Журнал неврологии и психиатрии им. C.C. Корсакова. Общество с ограниченной ответственностью Издательство Медиа Сфера, 2016. Vol. 116, № 9–2. P. 61–66.
320. Guerrini R. Et al. A randomized phase III trial of adjunctive zonisamide in pediatric patients with partial epilepsy // Epilepsia. Epilepsia, 2013. Vol. 54, № 8. P. 1473–1480.
321. Brigo F., Igwe S.C. Ethosuximide, sodium valproate or lamotrigine for absence seizures in children and adolescents // Cochrane Database of Systematic Reviews. John Wiley and Sons Ltd, 2017. Vol. 2017, № 2.
322. Wallace S.J. Myoclonus and epilepsy in childhood: A review of treatment with valproate, ethosuximide, lamotrigine and zonisamide // Epilepsy Res. Epilepsy Res, 1998. Vol. 29, № 2. P. 147–154.
323. Krauss G.L. et al. Randomized phase III study 306 Adjunctive perampanel for refractory partial-onset seizures // Neurology. Lippincott Williams and Wilkins, 2012. Vol. 78, № 18. P. 1408–1415.
324. French J.A. et al. Perampanel for tonic-clonic seizures in idiopathic generalized epilepsy. A randomized trial // Neurology. Lippincott Williams and Wilkins, 2015. Vol. 85, № 11. P. 950–957.
325. Fogarasi A. et al. Open-label study to investigate the safety and efficacy of adjunctive perampanel in pediatric patients (4 to <12 years) with inadequately controlled focal seizures or generalized tonic-clonic seizures // Epilepsia. Blackwell Publishing Inc., 2020. Vol. 61, № 1. P. 125–137.
326. Farkas V. et al. Efficacy and tolerability of adjunctive lacosamide in pediatric patients with focal seizures // Neurology. Neurology, 2019. Vol. 93, № 12. P. E1212–E1226.
327. Chadwick D.W. et al. A double-blind trial of gabapentin monotherapy for newly diagnosed partial seizures // Neurology. Lippincott Williams and Wilkins, 1998. Vol. 51, № 5. P. 1282–1288.
328. Panebianco M. et al. Gabapentin add-on treatment for drug-resistant focal epilepsy // Cochrane Database of Systematic Reviews. John Wiley and Sons Ltd, 2018. Vol. 2018, № 10.
329. Klein P. et al. A randomized, double-blind, placebo-controlled, multicenter, parallel-group study to evaluate the efficacy and safety of adjunctive brivaracetam in adult patients with uncontrolled partial-onset seizures // Epilepsia. Blackwell Publishing Inc., 2015. Vol. 56, № 12. P. 1890–1898.
330. Bresnahan R. et al. Clobazam add-on therapy for drug-resistant epilepsy // Cochrane Database of Systematic Reviews. John Wiley and Sons Ltd, 2019. Vol. 2019, № 10.
331. Ng Y.T. et al. Randomized, phase III study results of clobazam in Lennox-Gastaut syndrome // Neurology. Lippincott Williams and Wilkins, 2011. Vol. 77, № 15. P. 1473–1481.
332. Sofou K, Kristjánsdóttir R, Papachatzakis NE, Ahmadzadeh A, Uvebrant P.J Management of prolonged seizures and status epilepticus in childhood: a systematic review. Child Neurol. 2009 Aug;24(8):918-26. doi: 10.1177/0883073809332768. Epub 2009 Mar 30
333. Wolf P. Acute Drug Administration in Epilepsy: A Review // CNS Neurosci. Ther. CNS Neurosci Ther, 2011. Vol. 17, № 5. P. 442–448.
334. Brigo F. et al. Nonintravenous midazolam versus intravenous or rectal diazepam for the treatment of early status epilepticus: A systematic review with meta-analysis // Epilepsy Behav. Epilepsy Behav, 2015. Vol. 49. P. 325–336.
335. Pavlidou E., Tzitiridou M., Panteliadis C. Effectiveness of intermittent diazepam prophylaxis in febrile seizures: Long-term prospective controlled study // J. Child Neurol. J Child Neurol, 2006. Vol. 21, № 12. P. 1036–1040.
336. McIntyre J. et al. Safety and efficacy of buccal midazolam versus rectal diazepam for emergency treatment of seizures in children: A randomised controlled trial // Lancet. Elsevier B.V., 2005. Vol. 366, № 9481. P. 205–210.
337. Li S. et al. Prednisolone/prednisone as adrenocorticotropic hormone alternative for infantile spasms: a meta-analysis of randomized controlled trials // Developmental Medicine and Child Neurology. Blackwell Publishing Ltd, 2020. Vol. 62, № 5. P. 575–580.
338. Rajpurohit M. et al. Safety, Feasibility and Effectiveness of Pulse Methylprednisolone Therapy in Comparison with Intramuscular Adrenocorticotropic Hormone in Children with West Syndrome // Indian J. Pediatr. Springer, 2020.
339. Haberlandt E. et al. Adrenocorticotropic Hormone versus Pulsatile Dexamethasone in the Treatment of Infantile Epilepsy Syndromes // Pediatr. Neurol. Pediatr Neurol, 2010. Vol. 42, № 1. P. 21–27.
340. Прыгунова Т.М. Синдром Веста: отдаленные исходы в зависимости от этиологии и лечения (обзор литературы) // Русский журнал детской неврологии. Общество с ограниченной ответственностью «Издательский дом «АБВ-пресс», 2018. Vol. 13, № 4.
341. Coughlin CR 2nd, Tseng LA, Abdenur JE, Ashmore C, Boemer F, Bok LA, Boyer M, Buhas D, Clayton PT, Das A, Dekker H, Evangeliou A, Feillet F, Footitt EJ, Gospe SM Jr, Hartmann H, Kara M, Kristensen E, Lee J, Lilje R, Longo N, Lunsing RJ, Mills P, Papadopoulou MT, Pearl PL, Piazzon F, Plecko B, Saini AG, Santra S, Sjarif DR, Stockler-Ipsiroglu S, Striano P, Van Hove JLK, Verhoeven-Duif NM, Wijburg FA, Zuberi SM, van Karnebeek CDM. Consensus guidelines for the diagnosis and management of pyridoxine-dependent epilepsy due to α-aminoadipic semialdehyde dehydrogenase deficiency. J Inherit Metab Dis. 2021 Jan;44(1):178-192. doi: 10.1002/jimd.12332. Epub 2020 Dec 1. PMID: 33200442.
342. Шалькевич Л.В., Шанько Г.Г., Кот Д.А. Кортикостероидная терапия фармакорезистентных эпилепсий у детей // Международный неврологический журнал. ФЛП «ЗаславскийАлександрЮрьевич», 2014. № 3 (65). P. 20–27.
343. deVries E.E. et al. Inflammatory mediators in human epilepsy: A systematic review and meta-analysis // Neuroscience and Biobehavioral Reviews. ElsevierLtd, 2016. Vol. 63. P. 177–190.
344. Белоусова Е.Д., Ермаков A.Ю. Эпилептическая энцефалопатия с продолженной спайк-волновой активностью во сне: обзор литературы // Журнал неврологии и психиатрии им. ССКорсакова. Спецвыпуски. Издательство ’МедиаСфера’, 2014. Vol. 114, № 4. P. 52–58.
345. IizukaT, KanazawaN, KanekoJ, TominagaN, NonodaY, HaraA, OnozawaY, AsariH, HataT, KanekoJ, YoshidaK, SugiuraY, UgawaY, WatanabeM, TomitaH, KosakaiA, KanekoA, IshimaD, KitamuraE, NishiyamaK. CryptogenicNORSE: Itsdistinctiveclinicalfeaturesandresponsetoimmunotherapy. Neurol Neuroimmunol Neuroinflamm. 2017 Sep 25;4(6):e396. doi: 10.1212/NXI.0000000000000396. PMID: 28959704; PMCID: PMC5614728.
346. Белоусова Е.Д., Яблонская М.И., Тагирова М.К. и соавт. Иммунообусловленные эпилепсии у детей // Российский вестник перинатологии и педиатрии, 2015. Vol. 60, № 5. P. 26–32.
347. NabboutR. еt al. Efficacy of ketogenic diet in severe refractory status epilepticus initiating fever induced refractory epileptic encephalopathy in school age children (FIRES) // Epilepsia. Epilepsia, 2010. Vol. 51, № 10. P. 2033–2037.
348. Bello-Espinosa LE, Rajapakse T, Rho JM, Buchhalter J. Efficacy of intravenous immunoglobulin in a cohort of children with drug-resistant epilepsy. Pediatr Neurol. 2015 May;52(5):509-16. doi: 10.1016/j.pediatrneurol.2014.11.011. Epub 2015 Feb 13. PMID: 25882078.
349. Rezaei S. et al. Short-term and long-term efficacy of classical ketogenic diet and modified Atkins diet in children and adolescents with epilepsy: A systematic review and meta-analysis // Nutritional Neuroscience. Taylor and Francis Ltd., 2019. Vol. 22, № 5. P. 317–334.
350. Englot D.J., Chang E.F., Auguste K.I. Vagus nerve stimulation for epilepsy: A meta-analysis of efficacy and predictors of response - A review // Journal of Neurosurgery. J Neurosurg, 2011. Vol. 115, № 6. P. 1248–1255.
351. Maragkos G.A. et al. Quality of Life after Epilepsy Surgery in Children: A Systematic Review and Meta-Analysis // Clinical Neurosurgery. Oxford University Press, 2019. Vol. 85, № 6. P. 741–749.
352. Tomson T. et al. Executive Summary: Management of epilepsy in pregnancy: A report from the International League Against Epilepsy Task Force on Women and Pregnancy // Epilepsia. Blackwell Publishing Inc., 2019. Vol. 60, № 12. P. 2343–2345.
353. Thomas S. V., Syam U., Devi J.S. Predictors of seizures during pregnancy in women with epilepsy // Epilepsia. 2012. Vol. 53, № 5.
354. Vajda F.J.E. et al. Seizure control in antiepileptic drug-treated pregnancy // Epilepsia. Blackwell Publishing Inc., 2008. Vol. 49, № 1. P. 172–176.
355. Battino D. et al. Seizure control and treatment changes in pregnancy: Observations from the EURAP epilepsy pregnancy registry // Epilepsia. Epilepsia, 2013. Vol. 54, № 9. P. 1621–1627.
356. Hiilesmaa V.K., Teramo K.A. Fetal and Maternal Risks with Seizures // Epilepsy in Women. John Wiley and Sons, 2013. P. 115–127.
357. Edey S., Moran N., Nashef L. SUDEP and epilepsy-related mortality in pregnancy // Epilepsia. Blackwell Publishing Inc., 2014. Vol. 55, № 7.
358. EURAP - International Registry of Antiepileptic Drugs and Pregnancy [Electronic resource]. URL: https://eurapinternational.org/ (accessed: 30.05.2021).
359. Tomson T., Battino D., Perucca E. Teratogenicity of antiepileptic drugs // Curr. Opin. Neurol. Lippincott Williams and Wilkins, 2019. Vol. 32, № 2. P. 246–252.
360. Blotière P.O. et al. Risks of 23 specific malformations associated with prenatal exposure to 10 antiepileptic drugs // Neurology. Lippincott Williams and Wilkins, 2019. Vol. 93, № 2. P. E167–E180.
361. Карлов В.А. et al. Эпилепсия и беременность // Эпилепсия у детей и взрослых женщин и мужчин. Второе издание / ed. Карлов В.А. Москва: Издательство БИНОМ, 2019. P. 672–690.
362. Карлов В.А., Бурд С.Г., Михаловска-Карлова Е.П. Современность и эпилепсия. Современные и биоэтические аспекты эпилепсии. // Эпилепсия у детей и взрослых женщин и мужчин». Руководство для врачей. Второе издание. / ed. Карлов В.А. Москва: Издательство БИНОМ, 2019. P. 868–891.
363. Власов П.Н. Применение противоэпилептических препаратов во время беременности // Фармакотерапия отдельных состояний при беременности / ed. Володин Н.Н., Белоусов Ю.Б., Зырянов С.К. Москва: Миклош, 2012. P. 127–137.
364. Pariente G. Et al. Pregnancy Outcomes Following In Utero Exposure to Lamotrigine: A Systematic Review and Meta-Analysis // CNS Drugs. Adis, 2017. Vol. 31, № 6. P. 439–450.
365. Veroniki A.A. et al. Comparative safety of anti-epileptic drugs during pregnancy: A systematic review and network meta-analysis of congenital malformations and prenatal outcomes // BMC Med. BioMed Central Ltd., 2017. Vol. 15, № 1.
366. Morrow J.I. et al. Folic acid use and major congenital malformations in offspring of women with epilepsy: A prospective study from the UK Epilepsy and pregnancy register // J. Neurol. Neurosurg. Psychiatry. J Neurol Neurosurg Psychiatry, 2009. Vol. 80, № 5. P. 506–511.
367. Tomson T. et al. Comparative risk of major congenital malformations with eight different antiepileptic drugs: a prospective cohort study of the EURAP registry // Lancet Neurol. Lancet Publishing Group, 2018. Vol. 17, № 6. P. 530–538.
368. Meador K.J. et al. Fetal antiepileptic drug exposure and cognitive outcomes at age 6 years (NEAD study): A prospective observational study // Lancet Neurol. Lancet Neurol, 2013. Vol. 12, № 3. P. 244–252
369. Minicucci F., Ferlisi M., Brigo F., Mecarelli O., Meletti S., Aguglia U., Michelucci R., Mastrangelo M., Specchio N., Sartori S., Tinuper P. Management of status epilepticus in adults. Position paper of the Italian League against Epilepsy. Epilepsy & Behavior 102 (2020)
370. Kilbride R.D., Reynolds A.S., Szaflarski J.P., Hirsch L.J. Clinical outcomes following prolonged refractory status epilepticus (PRSE). Neurocrit Care 2013; 18 (3):374-385.
371. Özkara Ç. et al. Surgical outcome of epilepsy patients evaluated with a noninvasive protocol // Epilepsia. Lippincott Williams and Wilkins, 2000. Vol. 41, № SUPPL. 4.
372. Jette N., Reid A.Y., Wiebe S. Surgical management of epilepsy // CMAJ. Canadian Medical Association, 2014. Vol. 186, № 13. P. 997–1004.
373. Wiebe S., Jette N. Pharmacoresistance and the role of surgery in difficult to treat epilepsy // Nat. Rev. Neurol. Nat Rev Neurol, 2012. Vol. 8, № 12. P. 669–677.
374. Spencer S., Huh L. Outcomes of epilepsy surgery in adults and children // Lancet Neurol. Lancet Neurol, 2008. Vol. 7, № 6. P. 525–537.
375. Spencer S.S. et al. Health-related quality of life over time since resective epilepsy surgery // Ann. Neurol. Ann Neurol, 2007. Vol. 62, № 4. P. 327–334.
376. Ryvlin P., Cross J.H., Rheims S. Epilepsy surgery in children and adults // Lancet Neurol. Lancet Publishing Group, 2014. Vol. 13, № 11. P. 1114–1126.
377. Velasco A.L. et al. Electrical stimulation of the hippocampal epileptic foci for seizure control: A double-blind, long-term follow-up study // Epilepsia. Epilepsia, 2007. Vol. 48, № 10. P. 1895–1903.
378. Neligan A. et al. A survey of adult and pediatric epilepsy surgery in the United Kingdom // Epilepsia. Epilepsia, 2013. Vol. 54, № 5.
379. Niemeyer P. The transventricular amygdalohippocampectomy in temporal lobe epilepsy // Temporal lobe epilepsy. Charles C Thomas, 1958. P. 461–482.
380. Vakharia V.N. et al. Getting the best outcomes from epilepsy surgery // Ann. Neurol. John Wiley and Sons Inc., 2018. Vol. 83, № 4. P. 676–690.
381. Crandall P.H. Standard en bloc anterior temporal lobectomy // Surgery for epilepsy. Blackwell Scientific Publications, Boston, 1991. P. 118–129.
382. Wyler A.R. Anterior temporal lobectomy // Surg. Neurol. Surg Neurol, 2000. Vol. 54, № 5. P. 341–345.
383. Wen H.T. et al. Microsurgical anatomy of the temporal lobe: Part 1: Mesial temporal lobe anatomy and its vascular relationships as applied to amygdalohippocampectomy // Neurosurgery. Lippincott Williams and Wilkins, 1999. Vol. 45, № 3. P. 549–592.
384. Spencer D.D. et al. Access to the posterior medial temporal lobe structures in the surgical treatment of temporal lobe epilepsy // Neurosurgery. Neurosurgery, 1984. Vol. 15, № 5. P. 667–671.
385. Noachtar S., Borggraefe I. Epilepsy surgery: A critical review // Epilepsy Behav. Epilepsy Behav, 2009. Vol. 15, № 1. P. 66–72.
386. Connor D.E. et al. Vagal nerve stimulation for the treatment of medically refractory epilepsy: A review of the current literature // Neurosurg. Focus. Neurosurg Focus, 2012. Vol. 32, № 3.
387. Morris G.L. et al. Evidence-based guideline update: Vagus nerve stimulation for the treatment of epilepsy: Report of the guideline development subcommittee of the american academy of neurology // Neurology. Neurology, 2013. Vol. 81, № 16. P. 1453–1459.
388. Manuals « VNS Therapy « Healthcare Professionals, on VNS Therapy for Epilepsy | Cyberonics US [Electronic resource]. URL: https://dynamic.cyberonics.com/manuals/index_iframe_test.asp?lang=English-US (accessed: 30.05.2021).
389. Josephson C.B. et al. Systematic review and meta-analysis of standard vs selective temporal lobe epilepsy surgery // Neurology. Neurology, 2013. Vol. 80, № 18. P. 1669–1676.
390. Morrell F., Whisler W.W., Bleck T.P. Multiple subpial transection: A new approach to the surgical treatment of focal epilepsy // J. Neurosurg. J Neurosurg, 1989. Vol. 70, № 2. P. 231–239.
391. Spencer S.S. et al. Multiple subpial transection for intractable partial epilepsy: An international meta-analysis // Epilepsia. Epilepsia, 2002. Vol. 43, № 2. P. 141–145.
392. Hufnagel A. et al. Multiple subpial transection for control of epileptic seizures: Effectiveness and safety // Epilepsia. Epilepsia, 1997. Vol. 38, № 6. P. 678–688.
393. Gates J.R. et al. Response of Multiple Seizure Types to Corpus Callosum Section // Epilepsia. Epilepsia, 1987. Vol. 28, № 1. P. 28–34.
394. Wilson D.H., Reeves A.G., Gazzaniga M.S. “Central” commissurotomy for intractable generalized epilepsy: Series two // Neurology. Neurology, 1982. Vol. 32, № 7. P. 687–697.
395. Wyllie E. Catastrophic epilepsy in infants and children: Identification of surgical candidates // Epileptic Disord. Epileptic Disord, 1999. Vol. 1, № 4. P. 261–264.
396. Carreño M. et al. Intractable epilepsy in vascular congenital hemiparesis: Clinical features and surgical options // Neurology. Lippincott Williams and Wilkins, 2002. Vol. 59, № 1. P. 129–131.
397. González-Martínez J.A. et al. Hemispherectomy for catastrophic epilepsy in infants // Epilepsia. Epilepsia, 2005. Vol. 46, № 9. P. 1518–1525.
398. Elsharkawy A.E. et al. Outcome of extratemporal epilepsy surgery experience of a single center // Neurosurgery. Neurosurgery, 2008. Vol. 63, № 3. P. 516–525.
399. Крылов В.В.Хирургия эпилепсии. АБВ-пресс, 2019.
400. Bauer S., Hamer H.M. Extratemporal epilepsies // Handbook of Clinical Neurology. Elsevier B.V., 2012. Vol. 107. P. 241–256.
401. Greiner H.M. et al. Preresection intraoperative electrocorticography (ECoG) abnormalities predict seizure-onset zone and outcome in pediatric epilepsy surgery // Epilepsia. Blackwell Publishing Inc., 2016. Vol. 57, № 4. P. 582–589.
402. Kaufmann E. et al. European Expert Opinion on ANT-DBS therapy for patients with drug-resistant epilepsy (a Delphi consensus) // Seizure. W.B. Saunders Ltd, 2020. Vol. 81. P. 201–209.
403. Salanova V. et al. Long-term efficacy and safety of thalamic stimulation for drug-resistant partial epilepsy // Neurology. Lippincott Williams and Wilkins, 2015. Vol. 84, № 10. P. 1017–1025.
404. Parent M., Parent A. Single-axon tracing and three-dimensional reconstruction of centre médian-parafascicular thalamic neurons in primates // J. Comp. Neurol. J Comp Neurol, 2005. Vol. 481, № 1. P. 127–144.
405. Handforth A., DeSalles A.A.F., Krahl S.E. Deep brain stimulation of the subthalamic nucleus as adjunct treatment for refractory epilepsy // Epilepsia. Epilepsia, 2006. Vol. 47, № 7. P. 1239–1241.
406. Wellmer J., Voges J., Parpaley Y. Lesion guided radiofrequency thermocoagulation (L-RFTC) for hypothalamic hamartomas, nodular heterotopias and cortical dysplasias: Review and perspective // Seizure. W.B. Saunders Ltd, 2016. Vol. 41. P. 206–210.
407. Kameyama S. et al. MRI-guided stereotactic radiofrequency thermocoagulation for 100 hypothalamic hamartomas // J. Neurosurg. American Association of Neurological Surgeons, 2016. Vol. 124, № 5. P. 1503–1512.
408. Catenoix H. et al. SEEG-guided thermocoagulations: A palliative treatment of nonoperable partial epilepsies // Neurology. Lippincott Williams and Wilkins, 2008. Vol. 71, № 21. P. 1719–1726.
409. Gopinath S. et al. Seizure outcome following primary motor cortex-sparing resective surgery for perirolandic focal cortical dysplasia // Int. J. Surg. Elsevier Ltd, 2016. Vol. 36, № Pt B. P. 466–476.
410. Mula M., Sander J.W. Current and emerging drug therapies for the treatment of depression in adults with epilepsy // Expert Opinion on Pharmacotherapy. Taylor and Francis Ltd, 2019. Vol. 20, № 1. P. 41–45.
411. Orjuela-Rojas J.M. et al. Treatment of depression in patients with temporal lobe epilepsy: A pilot study of cognitive behavioral therapy vs. selective serotonin reuptake inhibitors // Epilepsy Behav. Academic Press Inc., 2015. Vol. 51. P. 176–181.
412. Li W., Ma D. A randomized controlled trial to evaluate the efficacy of paroxetine and doxepin in treating epileptic patients with depression // Zhong Linch Kangfu. 2005. Vol. 9. P. 20–21.
413. Усюкина М.В. Систематика и принципы терапии депрессивных расстройств при эпилепсии// Нервно-мышечные болезни. 2015. №4. URL: https://cyberleninka.ru/article/n/sistematika-i-printsipy-terapii-depressivnyh-rasstroystv-pri-epilepsii (дата обращения: 16.07.2021).
414. Hovorka J., Herman E., Nemcová I. Treatment of interictal depression with citalopram in patients with epilepsy // Epilepsy Behav. Epilepsy Behav, 2000. Vol. 1, № 6. P. 444–447.
415. Specchio L.M. et al. Citalopram as treatment of depression in patients with epilepsy // Clin. Neuropharmacol. Clin Neuropharmacol, 2004. Vol. 27, № 3. P. 133–136.
416. Kühn K.U. et al. Antidepressive treatment in patients with temporal lobe epilepsy and major depression: A prospective study with three different antidepressants // Epilepsy Behav. Academic Press Inc., 2003. Vol. 4, № 6. P. 674–679.
417. Gilliam F.G. et al. A Trial of Sertraline or Cognitive Behavior Therapy for Depression in Epilepsy // Ann. Neurol. John Wiley and Sons Inc., 2019. Vol. 86, № 4. P. 552–560.
418. Kanner A.M., Kozak A.M., Frey M. The use of sertraline in patients with epilepsy: Is it safe? // Epilepsy Behav. Epilepsy Behav, 2000. Vol. 1, № 2. P. 100–105.
419. Zhu S.Q., Luo L.J., YaX G. Short-Term efficacy of venlafaxine treating the depression in epilepsy patients // Chin J Rehabil. 2004. Vol. 19. P. 101.
420. Носов С.Г. Депрессия и эпилепсия: общие патогенетические закономерности развития и особенности лечения (обзор литературы) //Український вісник психоневрології. – 2012. – Т. 20. – №. 4. – С. 92-
421. Robertson M.M., Trimble M.R. The treatment of depression in patients with epilepsy A double-blind trial // J. Affect. Disord. J Affect Disord, 1985. Vol. 9, № 2. P. 127–136.
422. Alper K. et al. Seizure Incidence in Psychopharmacological Clinical Trials: An Analysis of Food and Drug Administration (FDA) Summary Basis of Approval Reports // Biol. Psychiatry. Biol Psychiatry, 2007. Vol. 62, № 4. P. 345–354.
423. Italiano D., Spina E., De Leon J. Pharmacokinetic and pharmacodynamic interactions between antiepileptics and antidepressants // Expert Opin. Drug Metab. Toxicol. Informa Healthcare, 2014. Vol. 10, № 11. P. 1457–1489.
424. Unterecker S. et al. Interaction of valproic acid and the antidepressant drugs doxepin and venlafaxine: Analysis of therapeutic drug monitoring data under naturalistic conditions // Int. Clin. Psychopharmacol. Lippincott Williams and Wilkins, 2014. Vol. 29, № 4. P. 206–211.
425. Hemeryck A., Belpaire F. Selective Serotonin Reuptake Inhibitors and Cytochrome P-450 Mediated Drug-Drug Interactions: An Update // Curr. Drug Metab. Bentham Science Publishers Ltd., 2002. Vol. 3, № 1. P. 13–37.
426. Berigan T., Harazin J. A sertraline/valproic acid drug interaction // Int. J. Psychiatry Clin. Pract. Martin Dunitz Ltd, 1999. Vol. 3, № 4. P. 287–288.
427. Macrodimitris S. et al. Group cognitive-behavioral therapy for patients with epilepsy and comorbid depression and anxiety // Epilepsy Behav. Epilepsy Behav, 2011. Vol. 11, № 1. P. 83–88.
428. Gandy M. et al. A feasibility trial of an Internet-delivered and transdiagnostic cognitive behavioral therapy treatment program for anxiety, depression, and disability among adults with epilepsy // Epilepsia. Blackwell Publishing Inc., 2016. Vol. 57, № 11. P. 1887–1896.
429. Пешеходько Д. И. Депрессия и тревожность при детской эпилепсии: обзорный анализ / Д.И. Пешеходько, Б.Д. Абдулазизов, А.А. Некишева // Перспективы науки. – 2020. – № 12(135). – С. 206-208.
430. Mula M. Pharmacological treatment of anxiety disorders in adults with epilepsy // Expert Opin. Pharmacother. Taylor and Francis Ltd, 2018. Vol. 19, № 17. P. 1867–1874.
431. Mula M. Treatment of anxiety disorders in epilepsy: An evidence-based approach // Epilepsia. Epilepsia, 2013. Vol. 54, № SUPPL. 1. P. 13–18.
432. Sepúlveda C. et al. Palliative care: The world health organization’s global perspective // J. Pain Symptom Manage. Elsevier Inc., 2002. Vol. 24, № 2. P. 91–96.
433. Pastrana T. et al. A matter of definition - Key elements identified in a discourse analysis of definitions of palliative care // Palliat. Med. Palliat Med, 2008. Vol. 22, № 3. P. 222–232.
434. Glauser T. et al. Evidence-based guideline: Treatment of convulsive status epilepticus in children and adults: Report of the guideline committee of the American epilepsy society // Epilepsy Currents. American Epilepsy Society, 2016. Vol. 16, № 1. P. 48–61.
435. Kossoff EH, Zupec-Kania BA, Auvin S, et al. Optimal clinical management of children receiving dietary therapies for epilepsy: Updated recommendations of the International Ketogenic Diet Study Group. Epilepsia Open. 2018;3(2):175-192. Published 2018 May 21. doi:10.1002/epi4.12225
436. D"Andrea Meira I, Romão TT, Pires do Prado HJ, Krüger LT, Pires MEP, da Conceição PO. Ketogenic Diet and Epilepsy: What We Know So Far. Front Neurosci. 2019;13:5. Published 2019 Jan 29. doi:10.3389/fnins.2019.00005
437. Vancampfort D., Ward P.B., Stubbs B. Physical fitness levels and moderators in people with epilepsy: A systematic review and meta-analysis // Epilepsy Behav. Academic Press Inc., 2019. Vol. 99.
438. Vancampfort D., Ward P.B., Stubbs B. Physical activity and sedentary levels among people living with epilepsy: A systematic review and meta-analysis // Epilepsy Behav. Academic Press Inc., 2019. Vol. 99.
439. Deng B.W. et al. A Meta-analysis of the Effectiveness of Acupuncture in the Treatment of Epilepsy // Zhen ci yan jiu = Acupunct. Res. NLM (Medline), 2018. Vol. 43, № 4. P. 263–268.
440. Nakken K.O., Brodtkorb E., Koht J. Epilepsi og rehabilitering // Tidsskr. Den Nor. legeforening. 2007.
441. Helde G. et al. An easily performed group education programme for patients with uncontrolled epilepsy - A pilot study // Seizure. W.B. Saunders Ltd, 2003. Vol. 12, № 7. P. 497–501.
442. Helde G. et al. A structured, nurse-led intervention program improves quality of life in patients with epilepsy: A randomized, controlled trial // Epilepsy Behav. Epilepsy Behav, 2005. Vol. 7, № 3. P. 451–457.
443. Michaelis R. et al. Psychological treatments for people with epilepsy // Cochrane Database Syst. Rev. John Wiley and Sons Ltd, 2017. Vol. 2017, № 10.
444. Haut S.R., Gursky J.M., Privitera M. Behavioral interventions in epilepsy // Curr. Opin. Neurol. Lippincott Williams and Wilkins, 2019. Vol. 32, № 2. P. 227–236.
445. Schröder J. et al. Efficacy of a psychological online intervention for depression in people with epilepsy: A randomized controlled trial // Epilepsia. Epilepsia, 2014. Vol. 55, № 12. P. 2069–2076.
446. Meyer B. et al. Effects of an epilepsy-specific Internet intervention (Emyna) on depression: Results of the ENCODE randomized controlled trial // Epilepsia. Epilepsia, 2019. Vol. 60, № 4. P. 656–668.
447. Tang V., Michaelis R., Kwan P. Psychobehavioral therapy for epilepsy // Epilepsy Behav. Epilepsy Behav, 2014. Vol. 32. P. 147–155.
448. Caller T.A. et al. A cognitive behavioral intervention (HOBSCOTCH) improves quality of life and attention in epilepsy // Epilepsy Behav. Epilepsy Behav, 2016. Vol. 57, № Pt A. P. 111–117.
449. Казенных Т.В., Бохан Н.А. Роль психотерапевтической коррекции при проведении комплексной терапии у больных пароксизмальными состояниями различного генеза // Международный журнал прикладных и фундаментальных исследований. Общество с ограниченной ответственностью" Издательский Дом" Академия …, 2016. № 6–3. P. 470–476.
450. Kertesz-BriestH.A. etal. Examining relations between neuropsychological and clinical epilepsy-specific factors with psychopathology and adaptive skills outcomes in youth with intractable epilepsy // Epilepsy Behav. EpilepsyBehav, 2020. Vol. 110.
451. Приказ Министерства здравоохранения Российской Федерации от 28 сентября 2020 г. N 1029н «Об утверждении перечней медицинских показаний и противопоказаний для санаторно-курортного лечения» http://publication.pravo.gov.ru/Document/View/0001202010270044.
452. WHO, ILAE, IBE. Epilepsy. A public health imperative. Geneva, 2019. xiii–xiii p.
453. The Lancet. Epilepsy prevention: an urgent global unmet need // Lancet. Lancet Publishing Group, 2019. Vol. 393, № 10191. P. 2564.
454. Приказ Министерства здравоохранения Российской Федерации от 24 декабря 2012 г. № 1404н “Об утверждении стандарта первичной медико-санитарной помощи при парциальной эпилепсии (фаза диагностики и подбора терапии)” [Electronicresource]. URL: https://minzdrav.gov.ru/documents/8345-prikaz-ministerstva-zdravoohraneniya-rossiyskoy-federatsii-ot-24-dekabrya-2012-g-1404n-ob-utverzhdenii-standarta-pervichnoy-mediko-sanitarnoy-pomoschi-pri-partsialnoy-epilepsii-faza-diagnostiki-i-podbora-terapii (accessed: 30.05.2021).
455. Приказ Министерства здравоохранения Российской Федерации от 24 декабря 2012 г. № 1439н “Об утверждении стандарта первичной медико-санитарной помощи при генерализованной эпилепсии” [Electronicresource]. URL: https://minzdrav.gov.ru/documents/8348-prikaz-ministerstva-zdravoohraneniya-rossiyskoy-federatsii-ot-24-dekabrya-2012-g-1439n-ob-utverzhdenii-standarta-pervichnoy-mediko-sanitarnoy-pomoschi-pri-generalizovannoy-epilepsii (accessed: 30.05.2021).
456. Приказ Министерства здравоохранения Российской Федерации от 24 декабря 2012 г.№ 1541н “Об утверждении стандарта специализированной медицинской помощи при эпилепсии” [Electronicresource]. URL: https://minzdrav.gov.ru/documents/8941-prikaz-ministerstva-zdravoohraneniya-rossiyskoy-federatsii-ot-24-dekabrya-2012-g-1541n-ob-utverzhdenii-standarta-spetsializirovannoy-meditsinskoy-pomoschi-pri-epilepsii.
457. Приказ Министерства здравоохранения Российской Федерации от 05.07.2016 № 468н “Об утверждении стандарта скорой медицинской помощи при судорогах, эпилепсии, эпилептическом статусе” [Electronicresource]. URL: http://publication.pravo.gov.ru/Document/View/0001201607180038.
458. Приказ Минздрава России от 10.12.2013 № 916н «О перечне видов высокотехнологичной медицинской помощи» [Electronicresource]. URL: https://minjust.consultant.ru/documents/8516.
459. Labiner D.M. et al. Essential services, personnel, and facilities in specialized epilepsy centers-Revised 2010 guidelines // Epilepsia. Epilepsia, 2010. Vol. 51, № 11. P. 2322–2333.
460. Benbadis S.R., Differential diagnosis of epilepsy: acritical review. Epilepsy Behav 15: 2009: 15–21
461. Wiglusz M.S., Landowski J, Michalak L, Cubała WJ. Validation of the Hospital Anxiety and Depression Scale in patients with epilepsy. Epilepsy Behav. 2016 May;58:97-101. doi: 10.1016/j.yebeh.2016.03.003. Epub 2016 Apr 9. PMID: 27064829.
462. Котов А.С. Сбор анамнеза и осмотр у пациентов с эпилепсией. Клиническая лекция / А.С. Котов, К.В. Фирсов // Русский медицинский журнал. Медицинское обозрение. – 2019. – Т. 3. – № 7. – С. 4-7.
463. Наумова Г. И. Организация медицинской помощи пациентам с впервые развившимся судорожным припадком // Неврология, нейропсихиатрия, психосоматика. 2009 (2): С. 37-41.
464. Yeshokumar AK, Coughlin A, Fastman J, Psaila K, Harmon M, Randell T, Schorr EM, Han H, Hoang H, Soudant C, Jette N. Seizures in autoimmune encephalitis-A systematic review and quantitative synthesis. Epilepsia. 2021 Feb;62(2):397-407.
465. Кочеткова О. Н. Нейроцистицеркоз: особенности клинико-патоморфологической картины, судорожного синдрома, современные методы диагностики и лечения / О.Н. Кочеткова, А.И. Кочетков // Вестник Воронежского государственного университета. Серия: Химия. Биология. Фармация. – 2013. – № 2. – С. 110-115.
466. Еремин, А.О. Клинические особенности эпилепсии у пациентов с ВИЧ-инфекцией / А.О. Еремин, И.Н. Тихомирова // Вестник Совета молодых учёных и специалистов Челябинской области. – 2016. – Т. 5. – № 4(15). – С. 27-29.
467. Фомина М.Ю., Щербук Ю.А., Воронин Е.Е., Рахманова А.Г. Особенности поражения нервной системы при перинатальной и парентеральной ВИЧ-инфекции / // Медико-биологические и социально-психологические проблемы безопасности в чрезвычайных ситуациях. – 2009. – № 2. – С. 55-61.
468. Krumholz A. et al. Evidence-based guideline: Management of an unprovoked first seizure in adults // Neurology. Lippincott Williams and Wilkins, 2015. Vol. 84, № 16. P. 1705–1713.
469. Koutroumanidis M. et al. The role of EEG in the diagnosis and classification of the epilepsy syndromes: a tool for clinical practice by the ILAE Neurophysiology Task Force (Part 2). EpilepticDisord. 2017 Dec 1;19(4):385-437. doi: 10.1684/epd.2017.0952. PMID: 29350182.
470. Перепелова Е.М, Перепелов В.А, Меркулова М.С, Синицын В.Е. Современные подходы к диагностике с помощью магнитно-резонансной томографии эпилептогенных и сопряженных с ними поражений головного мозга. Неврология, нейропсихиатрия, психосоматика. 2018;(спецвыпуск 1):4-11.
471. Wolf NI, Bast T, Surtees R. Epilepsy in inborn errors of metabolism. EpilepticDisord. 2005 Jun;7(2):67-81.
472. Tomson T, Battino D, Bromley R, Kochen S, Meador K, Pennell P, Thomas SV. Management of epilepsy in pregnancy: a report from the International League Against Epilepsy Task Force on Women and Pregnancy. Epileptic Disord. 2019 Dec 1;21(6):497-517.
473. French J.A. et al. Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study // Lancet. Lancet Publishing Group, 2016. Vol. 388, № 10056. P. 2153–2163.
474. Muthaffar OY. Neurosciences (Riyadh) Treating epilepsy with options otherthan antiepileptic medications. 2020 Aug;25(4):253-261. doi: 10.17712/nsj.2020.4.20200010.
475. Клинические рекомендации по диагностике и лечению эпилепсии. В кн. Детская неврология. Клинические рекомендации. Выпуск1. Под редакцией В.И. Гузевой – Москва: ООО «МК»: 2014; стр.316-317.
476. Белоусова, Е. Д. Гормональная терапия синдрома Веста / Е. Д. Белоусова, И. В. Шулякова, Т. Г. Охапкина // Журнал неврологии и психиатрии им. C.C. Корсакова. – 2016. – Т. 116. – № 9-2. – С. 61-66. – DOI 10.17116/jnevro20161169261-66
477. Haberlandt E, Weger C, Baumgartner Sigl S, et al. Adrenocorticotropic hormone versus pulsatile dexamethasone in the treatment of infantile epilepsy syndromes. PediatrNeurol. 2010;42:21–7.
478. К.Ю. Мухин, А.С. Петрухин. Эпилептические синдромы. Диагностика и стандарты терапии (Справочное руководство), М.; 2005; стр.75.
479. Chiron C, Dumas C, Jambaqué I, Mumford J, Dulac O. Randomized trial comparing vigabatrin and hydrocortisone in infantile spasms due to tuberous sclerosis. Epilepsy Res. 1997 Jan; 26 (2): 389-95. doi: 10.1016/s0920-1211(96)01006-6. PMID: 9095401.
480. Silbergleit R., Durkalski V., Lowenstein D., Conwit R., Pancioli A., Palesch Y., Barsan W. for the NETT Investigators. Intramuscular versus Intravenous Therapy for Prehospital Status Epilepticus. N Engl J Med 2012; 366: 591-600 DOI: 10.1056/NEJMoa1107494.
481. Berg A.T., Testa F.M., Levy S.R. Complete remission in nonsyndromic childhood-onset epilepsy // Ann. Neurol. Ann Neurol, 2011. Vol. 70, № 4. P. 566–573.
482. Goellner E. et al. Timing of early and late seizure recurrence after temporal lobe epilepsy surgery // Epilepsia. Epilepsia, 2013. Vol. 54, № 11. P. 1933–1941.
483. Lindsten H., Stenlund H., Forsgren L. Remission of seizures in a population-based adult cohort with a newly diagnosed unprovoked epileptic seizure // Epilepsia. Epilepsia, 2001. Vol. 42, № 8. P. 1025–1030.
484. Blume W.T. et al. Glossary of descriptive terminology for ictal semiology: Report of the ILAE Task Force on classification and terminology // Epilepsia. Epilepsia, 2001. Vol. 42, № 9. P. 1212–1218.
485. Berg A.T. et al. Revised terminology and concepts for organization of seizures and epilepsies: Report of the ILAE Commission on Classification and Terminology, 2005-2009 // Epilepsia. Epilepsia, 2010. Vol. 51, № 4. P. 676–685.
486. Zinchuk M, Kustov G, Pashnin E, et al. Validation of the Generalized Anxiety Disorder-7 (GAD-7) in Russian people with epilepsy. Epilepsy Behav. 2021; 123:108269.
487. Gaspard N., Hirsch L. J., Sculier Cl., Loddenkemper T., Baalen A. van, Lancrenon J., Emmery M., Specchio N., Farias-Moeller R., Wong N., Nabbout R. New-Onset Refractory Status Epilepticus –NORSE and Febrile Infection-Related Epilepsy Syndrome –FIRES: state of the art and perspectives. Epilepsia. 2018;1–8.
488. Prasad M, Krishnan PR, Sequeira R, Al-Roomi K. Anticonvulsant therapy for status epilepticus. Cochrane Database Syst Rev 2014; 9:CD003723. doi:10.1002/14651858.CD003723.pub3.
489. Chamberlain JM, Okada P, Holsti M, Mahajan P, Brown KM, Vance C, Gonzalez V, Lichenstein R, Stanley R, Brousseau DC, Grubenhoff J, et al. Lorazepam vs diazepam for pediatric status epilepticus: A randomized clinical trial. JAMA. 2014;311(16):1652–60
490. Silbergleit R, Durkalski V, Lowenstein D, Conwit R, Pancioli A, Palesch Y, Barsan W, Investigators N. Intramuscular versus intravenous therapy for prehospital status epilepticus. The New England journal of medicine. 2012;366(7):591–600 .
491. McIntyre J, Robertson S, Norris E, Appleton R, Whitehouse WP, Phillips B, Martland T, Berry K, Collier J, Smith S, Choonara I. Safety and efficacy of buccal midazolam versus rectal diazepam for emergency treatment of seizures in children: a randomised controlled trial. Lancet. 2005;366(9481):205–10.
492. Talukdar B, Chakrabarty B. Efficacy of buccal midazolam compared to intravenous diazepam in controlling convulsions in children: a randomized controlled trial. Brain Dev. 2009;31(10):744–9.
493. Scott RC, Besag FM, Neville BG. Buccal midazolam and rectal diazepam for treatment of prolonged seizures in childhood and adolescence: a randomised trial. Lancet. 1999;353(9153):623–6.
494. Mpimbaza A, Ndeezi G, Staedke S, Rosenthal PJ, Byarugaba J. Comparison of buccal midazolam with rectal diazepam in the treatment of prolonged seizures in ugandan children: a randomized clinical trial. Pediatrics. 2008;121(1):e58–64.
495. Trinka E, Höfler J, Zerbs A, Brigo F. Efficacy and safety of intravenous valproate for status epilepticus: a systematic review. CNS Drugs 2014;28:623-639
496. Agarwal P, Kumar N, Chandra R, Gupta G, Antony AR, Garg N. Randomized study of intravenous valproate and phenytoin in status epilepticus. Seizure. 2007;16(6):527–32
497. Malamiri RA, Ghaempanah M, Khosroshahi N, Nikkhah A, Bavarian B, Ashrafi MR. Efficacy and safety of intravenous sodium valproate versus phenobarbital in controlling convulsive status epilepticus and acute prolonged convulsive seizures in children: a randomised trial. Eur J Paediatr Neurol. 2012;16(5):536–41.
498. Mehta V, Singhi P, Singhi S. Intravenous sodium valproate versus diazepam infusion for the control of refractory status epilepticus in children: a randomized controlled trial. J Child Neurol. 2007;22(10):1191–7.
499. Hubert P., Parain D., Vallee L. Management of convulsive status epilepticus in infants and children. Rev Neurol (Paris). 2009 Apr;165(4):390-7.
500. Evidence-based guideline: treatment of convulsive status epilepticus in children and adults: report of the guideline committee of the American Epilepsy Society. Epilepsy Curr 2016;16:48-61.
501. Trinka Eu., Ho¨fler J., Leitinger M., Brigo F. Pharmacotherapy for Status Epilepticus Drugs (2015) 75:1499–1521.
502. Misra UK, Kalita J, Maurya PK. Levetiracetam versus lorazepam in status epilepticus: a randomized, open labeled pilot study. J Neurol. 2012;259(4):645–8
503. Glauser T, Shinnar S, Gloss D, Alldredge B, Arya R, Bainbridge J, et al. Evidence-based guideline: treatment of convulsive status epilepticus in children and adults: report of the guideline committee of the American Epilepsy Society. Epilepsy Curr 2016;16:48-61.
504. Zelano J, Kumlien E. Levetiracetam as alternative stage two antiepileptic drug in status epilepticus: a systematic review. Seizure 2012;21:233-236
505. Kima D., Jae-Moon Kima, Yong Won Chob , Kw Ik Yangc Dong Wook Kimd , Soon-Tae Leee Young Joo Nof , Jong-Geun Seog Jung-Ick Byunh Kyung Wook Kangi Keun Tae Kimb Antiepileptic Drug Therapy for Status Epilepticus J Clin Neurol 2021;17(1):11-19.
506. Vossler D. G., Bainbridge J. L., Boggs J. G., Novotny E. J., Loddenkemper T., Faught E., Amengual-Gual M., Fischer S. N., Gloss D. S., Olson D. M., Towne A. R., Naritoku D., Welty. T. E. Treatment of Refractory Convulsive Status Epilepticus: A Comprehensive Review by the American Epilepsy Society Treatments Committee. Epilepsy Currents 2020, Vol. 20(5) 245-264).
507. Santamarina E, González-Cuevas M, Toledo M, Jiménez M, Becerra JL, Quílez A, et al. Intravenous lacosamide (LCM) in status epilepticus (SE): weight-adjusted dose and efficacy. Epilepsy Behav 2018;84:93-98.
508. Ramsay RE, Sabharwal V, Khan F, Dave H, Kafai C, Shumate R, et al. Safety & pK of IV loading dose of lacosamide in the ICU. Epilepsy Behav 2015;49:340-342.
509. Shorvon S, Baulac M, Cross H, Trinka E, Walker M; Task Force on Status Epilepticus of the ILAE Commission for European Affairs. The drug treatment of status epilepticus in Europe: consensus document from a workshop at the first London Colloquium on Status Epilepticus. Epilepsia 2008;49:1277-1285.
510. Shorvon S, Ferlisi M. The treatment of super-refractory status epilepticus: a critical review of available therapies and a clinical treatment protocol. Brain 2011;134:2802-2818.
511. Claassen J, Hirsch LJ, Emerson RG, Mayer SA. Treatment of refractory status epilepticus with pentobarbital, propofol, or midazolam: a systematic review. Epilepsia 2002;43:146-153.
512. Rossetti AO, Milligan TA, Vulliémoz S, Michaelides C, Bertschi M, Lee JW. A randomized trial for the treatment of refractory status epilepticus. Neurocrit Care 2011;14:4-10.
513. Gomes D, Pimentel J, Bentes C, Aguiar de Sousa D, Antunes AP, Alvarez A, et al. Consensus protocol for the treatment of super-refractory status epilepticus. Acta Med Port 2018;31:598-605.
514.Fung LW, Yam KM; Yau M. LY. Ketamine use for super-refractory status epilepticus in children. Hong Kong Med J 2020 Dec;26(6):549–50.
515. Rosati A, L"Erario M, Ilvento L, Cecchi C, Pisano T, Mirabile L, et al. Efficacy and safety of ketamine in refractory status epilepticus in children. Neurology 2012;79:2355-2358.
516. Kramer U, Chi C-S, Lin K-L, et al. Febrile infection-related epilepsy syndrome (FIRES): pathogenesis, treatment, and outcome: a multicenter study on 77 children. Epilepsia. 2011;52:1956–65.
517. Hirsch LJ, Gaspard N, van Baalen A, et al. Proposed consensus definitions for new-onset refractory status epilepticus (NORSE), febrile infection-related epilepsy syndrome (FIRES), and related conditions. Epilepsia. 2018. https://doi.org/10.1111/epi.14016.
518. Khawaja AM, DeWolfe JL, Miller DW, Szaflarski JP. New-onset refractory status epilepticus (NORSE)–the potential role for immunotherapy. Epilepsy Behav. 2015;47:17–23.
519. Zeiler F.A., Matuszczak M., Teitebaum J., Kazina C.J., Gillman L.M.. Plasmopheresis for refractory status epilepticus Part I: a scoping systematic review of adult literature. Seizures,2016 43:14-22.
520. Zeiler F.A., Matuszczak M., Teitebaum J., Kazina C.J., Gillman L.M. Plasmopheresis for refractory status epilepticus Part II: a scoping systematic review of pediatric literature. Seizures, 2016 43:61-68..
521. Zeiler F A, Zeiler K J , Teitelbaum J , Gillman L M , West M. VNS for refractory status/Epilepsy Res 2015 May;112:100-13.
522. Dibue-Adjei M, Brigo Fr., Yamamoto T., Vonck Kr., Trinka Eu.. Vagus nerve stimulation in refractory and super-refractory status epilepticus e A systematic review/ Brain Stimulation 12 (2019) 1101e1110.
523. O"Connor SE , Ream MA, Richardson C, Mikati MA, Trescher WH, Byler DL et al. The ketogenic diet for the treatment of pediatric status epilepticus Pediatr Neurol. 2014 Jan;50(1):101-3.
524. Lai-wah Eva Fung The ketogenic diet for the treatment of pediatric status epilepticus Pediatr Neurol. 2014 Aug;51(2):e7. doi: 10.1016/j.pediatrneurol.2014.03.016.
525. Barkovich A.J., Guerrini R., Kuznetsk RI. et al. A developmental and genetic classification for malformations of cortical development: update 2012 // Brain. – 2012.- V.135.- P. 1348 – 1369.
526. Pearl P.L. Inherited metabolic epilepsies: the top 10 diagnoses you cannot afford to miss. // In: Inherited metabolic epilepsies / Eds. P.L. Pearl. – 2013. – DesmosMedical, N.Y. – P.1-13.
527. Hauser W.A. Epidemiology of epilepsy in children. // In: Eds.: J.M. Pellock, D.R. Nordli, Sankar R., Wheless J.W. / Pellock᾽s Pediatric Epilepsy. Diagnosis and their Therapy, 4th edition – DemosMedical, USA. – 2017. – P. 177 – 205.
528. Hsu WW et al. Systematic review and meta-analysis of the efficacy and safety of perampanel in the treatment of partial-onset epilepsy. CNS Drugs. 2013 Oct; 27(10):817-27. doi: 10.1007/s40263-013-0091-9. Erratum in: CNS Drugs. 2013 Dec;27(12):1143. PMID: 23918722; PMCID: PMC3784051.•
529. Steinhoff BJ et al. Efficacy and safety of adjunctive perampanel for the treatment of refractory partial seizures: a pooled analysis of three phase III studies. Epilepsia. 2013 Aug;54(8):1481-9. doi: 10.1111/epi.12212. Epub 2013 May 10. PMID: 23663001.
530. O"Callaghan FJ, Edwards SW, Alber FD, Hancock E, Johnson AL, Kennedy CR, Likeman M, Lux AL, Mackay M, Mallick AA, Newton RW, Nolan M, Pressler R, Rating D, Schmitt B, Verity CM, Osborne JP; participating investigators. Safety and effectiveness of hormonal treatment versus hormonal treatment with vigabatrin for infantile spasms (ICISS): a randomised, multicentre, open-label trial. Lancet Neurol. 2017 Jan;16(1):33-42. doi: 10.1016/S1474-4422(16)30294-0. Epub 2016 Nov 10. PMID: 27838190.
531. Chellamuthu P, Sharma S, Jain P, Kaushik JS, Seth A, Aneja S. High dose (4 mg/kg/day) versus usual dose (2 mg/kg/day) oral prednisolone for treatment of infantile spasms: an open-label, randomized controlled trial. Epilepsy Res. 2014 Oct;108(8):1378-84. doi: 10.1016/j.eplepsyres.2014.06.019. Epub 2014 Jul 5. PMID: 25048310.
532. Gaspard N, Hirsch LJ, Sculier C, Loddenkemper T, van Baalen A, Lancrenon J, Emmery M, Specchio N, Farias-Moeller R, Wong N, Nabbout R. New-onset refractory status epilepticus (NORSE) and febrile infection-related epilepsy syndrome (FIRES): State of the art and perspectives. Epilepsia. 2018 Apr;59(4):745-752. doi: 10.1111/epi.14022. Epub 2018 Feb 24. PMID: 29476535.
533. van Engelen B. G. M. et al. Immunoglobulin treatment in epilepsy, a review of the literature //Epilepsy research. – 1994. – Т. 19. – №. 3. – С. 181-190.
534. Бембеева Р.Ц., Заваденко Н.Н. Внутривенные иммуноглобулины в терапии аутоиммунных заболеваний нервной системы у детей. Журнал неврологии и психиатрии им. С.С. Корсакова. 2015;115(8):83-93.
535.Sourbron J. et al. Ketogenic diet for the treatment of pediatric epilepsy: review and meta-analysis //Child"s Nervous System. – 2020. – Т. 36. – №. 6. – С. 1099-1109.
536. Zi-yu Zhao, Hong-ying Wang, Bin Wen, Zhi-bo Yang, Kang Feng, Jing-chun Fan. A Comparison of Midazolam, Lorazepam, and Diazepam for the Treatment of Status Epilepticus in Children: A Network Meta-analysis. Journal of Child Neurology. 2017, 1-15;
537. Prasad M, Krishnan PR, Sequeira R, Al-Roomi K. Anticonvulsant therapy for status epilepticus. Cochrane Database of Systematic Reviews 2014, Issue 9. Art. No.: CD003723. DOI: 10.1002/14651858.CD003723.pub3).
538. D. G. Vossler, J. L. Bainbridge, J. G. Boggs, E. J. Novotny, T. Loddenkemper, E. Faught, M. Amengual-Gual, S. N. Fischer , D. S. Gloss, D. M. Olson, A. R. Towne, D. Naritoku, T. E. Welty. Treatment of Refractory Convulsive Status Epilepticus: A Comprehensive Review by the American Epilepsy Society Treatments Committee. Epilepsy Currents 2020, Vol. 20(5) 245-264.
539. D.G. Vossler, J.L. Bainbridge, J.G. Boggs, E.J. Novotny, T.Loddenkemper, E. Faught, M. Amengual-Gual, S.N. Fischer, D.S. Gloss, D.M. Olson, A.R. Towne, D. Naritoku, T. E. Welty. Treatment of Refractory Convulsive Status Epilepticus: A Comprehensive Review by the American Epilepsy Society Treatments Committee. Epilepsy Currents 2020, Vol. 20(5) 245-264;
540. Daeyoung Kima , Jae-Moon Kima Yong Won Chob , Kwang Ik Yangc Dong Wook Kimd , Soon-Tae Leee Young Joo Nof , Jong-Geun Seog Jung-Ick Byunh Kyung Wook Kangi Keun Tae Kimb. Antiepileptic Drug Therapy for Status Epilepticus. J Clin Neurol 2021;17(1):11-19.
541. Cetin Okuyaz, Kürşad Aydin, Kivilcim Gücüyener, Ayşe Serdaroğlu. Treatment of electrical status epilepticus during slow-wave sleep with high-dose corticosteroid. Pediatr Neurol. 2005 Jan;32(1):64-7; B.
542. van den Munckhof, Al. Arzimanoglou, E. Perucca, H. C. van Teeseling, F. S. S. Leijten, K. P. J. Braun, Fl. E. Jansen. Corticosteroids versus clobazam in epileptic encephalopathy with ESES: a European multicentre randomised controlled clinical trial (RESCUE ESES*) Trials 2020 Nov 23;21(1):957.
543.Клинические рекомендации «Алгоритмы специализированной медицинской помощи больным с сахарным диабетом»/ под ред. И.И. Дедова, М.В. Шестаковой, А.Ю Майорова. 10-й выпуск. 2021; 24(S1): С 70-71.
544. Клинические рекомендации по сердечно-легочной реанимации у детей. Российский национальный Совет по реанимации. Объединение детских анестезиологов реаниматологов. 2014г)
545. Shpak AA, Guekht AB, Druzhkova TA, Rider FK, Gulyaeva NV. Brain-derived neurotrophic factor in blood serum and lacrimal fluid of patients with focal epilepsy. Epilepsy Res. 2021 Oct; 176:106707. doi: 10.1016/j.eplepsyres.2021.106707. Epub 2021 Jun 29. PMID: 34225232.
546. Международная классификация болезней (10 пересмотр). Классификация психических и поведенческих расстройств. Санкт-Петербург, 1994; 300 с.
547. Базилевич С.Н., Прокудин М.Ю., Аверьянов Д.А., Дыскин Д.Е. Эпилептический статус: реальность 2021 // Известия Российской Военно-медицинской академии. - 2021. - Т. 40. - №4. - C. 59-68. doi: 10.17816/rmmar83623
548. M. Ngampoopun, P. Suwanpakdee, N. Jaisupa, Ch. Nabangchang. Effectiveness and Adverse Effect of Intravenous Lacosamide in Nonconvulsive Status Epilepticus and Acute Repetitive Seizures in Children. Neurol Res Int. 2018 Jun 10;2018:8432859.
doi: 10.1155/2018/8432859. eCollection 2018.
549. Arkilo D., Gustafson M., Ritter F. J. Clinical experience of intravenous lacosamide in infants and young children. European Journal of Paediatric Neurology. 2016;20(2):212–217. doi: 10.1016/j.ejpn.2015.12.013
550. Northam R.S., Hernandez A.W., Litzinger M.J., Minecan D.N., Glauser T.A., Mangat S., Zheng C., Souppart C., Sturm Y. Oxcarbazepine in infants and young children with partial seizures. Oxcarbazepine in infants and young children with partial seizures. Pediatr Neurol. 2005 Nov;33(5):337-44. doi: 10.1016/j.pediatrneurol.2005.05.011. PMID: 16243221.
551. Elterman R., Glauser T.A., Wyllie E., et al, and the Topiramate YP Study Group: A double blind, randomized trial of topiramate as adjunctive therapy for partial-onset seizures in children. Neurology 1999; 52:1338–1344.
552. de la Rosa J. S. O. et al. Efficacy of lacosamide in children and adolescents with drug-resistant epilepsy and refractory status epilepticus: A systematic review //Seizure. – 2018. – Т. 56. – С. 34-40.
553. Yorns Jr W. R. et al. Efficacy of lacosamide as adjunctive therapy in children with refractory epilepsy //Journal of child neurology. – 2014. – Т. 29. – №. 1. – С. 23-27.
554. Verhelst H. et al. Steroids in intractable childhood epilepsy: clinical experience and review of the literature //Seizure. – 2005. – Т. 14. – №. 6. – С. 412-421.
555. Kurian M., Korff C. M. Steroids in pediatric epilepsy: infantile spasms and beyond //Epileptologie. – 2011. – Т. 28. – №. 1. – С. 15-20.
Приложение А1. Состав рабочей группы по разработке и пересмотру клинических рекомендаций
Айвазян Сергей Оганесович, к.м.н, доценткафедрыневрологии детского возраста ГБОУ ДПО РМАПО Минздрава РФ, ведущий научный сотрудник Научно-практического центра медицинской помощи детям ДЗМ.
Акжигитов Ренат Гайясович, к.м.н., заместитель директора ГБУЗ «Научно-практический психоневрологический центр имени З.П. Соловьева» Департамента здравоохранения города Москвы.
Алферова Вера Вадимовна, д.м.н., главный научный сотрудник кафедры неврологии, нейрохирургии и медицинской генетики ФГАОУ ВО «Российский национальный исследовательский медицинский университет имени Н.И. Пирогова» Министерства здравоохранения Российской Федерации, член президиума Всероссийского общества неврологов.
Бадалян Оганес Левонович, д.м.н., профессор кафедры неврологии, нейрохирургии и медицинской генетики лечебного факультета ФГАОУ ВО «Российский национальный исследовательский медицинский университет имени Н.И. Пирогова» Министерства здравоохранения Российской Федерации
Базилевич Сергей Николаевич, к.м.н., доцент кафедры и клиники нервных болезней им. М.И. Аствацатурова ФГБВОУ ВО «Военно-медицинская академия им. С. М. Кирова» Министерства обороны Российской Федерации.
Батышева Татьяна Тимофеевна, д.м.н., профессор, Главный внештатный детский специалист невролог, Директор ГБУЗ «Научно-практический центр детской психоневрологии ДЗМ».
Белкин Андрей Августович, д.м.н., профессор кафедры физической и реабилитационной медицины федерального государственного бюджетного образовательного учреждения высшего образования "Уральский государственный медицинский университет" Министерства здравоохранения Российской Федерации, директор ООО «Клинический институт мозга», профессор, главный внештатный специалист УрФО РФ по медицинской реабилитации.
Белоусова Елена Дмитриевна, д.м.н., профессор ФГАОУ ВО «Российский национальный исследовательский медицинский университет имени Н.И. Пирогова» Министерства здравоохранения Российской Федерации, заведующая отделом психоневрологии и эпилептологии ОСП Научно-исследовательский клинический институт педиатрии им. Ю.Е. Вельтищева.
Бурд Сергей Георгиевич, д.м.н., профессор кафедры неврологии, нейрохирургии и медицинской генетики лечебного факультета ФГАОУ ВО «Российский национальный исследовательский медицинский университет имени Н.И. Пирогова» Министерства здравоохранения Российской Федерации.
Власов Павел Николаевич, д.м.н., профессор кафедры нервных болезней ФГБОУ ВО МГМСУ им. А.И.Евдокимова Министерства здравоохранения Российской Федерации, член президиума Всероссийского общества неврологов.
Гехт Алла Борисовна, д.м.н., профессор, директор ГБУЗ «Научно-практический психоневрологический центр имени З.П. Соловьева» Департамента здравоохранения города Москвы, профессор кафедры неврологии, нейрохирургии и медицинской генетики ФГАОУ ВО «Российский национальный исследовательский медицинский университет имени Н.И. Пирогова» Министерства здравоохранения Российской Федерации, вице-президент Международной противоэпилептической лиги, ученый секретарь Всероссийского общества неврологов.
Григорьева Елена Владимировна, д.м.н., ассистент кафедры лучевой диагностики, заведующая отделением лучевой диагностики КМЦ ФГБОУ ВО МГМСУ им. А.И.Евдокимова Министерства здравоохранения Российской Федерации.
Гузева Валентина Ивановна, д.м.н., профессор, член-корреспондент Российской Академии Естествознания, заведующая кафедрой неврологии, нейрохирургии и медицинской генетики ФГБОУ ВО Санкт-Петербургский государственный педиатрический медицинский университет Министерства здравоохранения Российской Федерации, главный внештатный детский специалист Министерства здравоохранения Российской Федерации.
Гузева Виктория Валентиновна, д.м.н., доцент, профессор кафедры неврологии, нейрохирургии и медицинской генетики ФГБОУ ВО Санкт-Петербургский государственный педиатрический медицинский университет Министерства здравоохранения Российской Федерации.
Гузева Оксана Валентиновна, д.м.н., доцент, профессор кафедры неврологии, нейрохирургии и медицинской генетики ФГБОУ ВО Санкт-Петербургский государственный педиатрический медицинский университет Министерства здравоохранения Российской Федерации.
Гуляева Наталия Валерьевна, д.б.н., профессор, заведующий лабораторией функциональной биохимии нервной системы Института высшей нервной деятельности и нейрофизиологии РАН, заведующая отделом изучения нейробиологических механизмов пограничных психических расстройств ГБУЗ НПЦ им. З.П.Соловьева ДЗМ.
Гусев Евгений Иванович, д.м.н., академик РАН, заведующий кафедрой неврологии, нейрохирургии и медицинской генетики ФГАОУ ВО «Российский национальный исследовательский медицинский университет имени Н.И. Пирогова» Министерства здравоохранения Российской Федерации, президент Национальной Ассоциации по борьбе с инсультом, Председатель Правления Всероссийского общества неврологов.
Дадали Елена Леонидовна, д.м.н., профессор, заведующая Научно-консультативным отделением ФГБНУ «Медико-генетический научный центр имени академика Н.П. Бочкова».
Дмитренко Диана Викторовна, д.м.н., профессор, заведующая кафедрой медицинской генетики и клинической нейрофизиологии ИПО ФГБОУ ВО КрасГМУ им. проф. В.Ф. Войно-Ясенецкого, руководитель неврологического центра университетской клиники ФГБОУ ВО КрасГМУ им. проф. В.Ф. Войно-Ясенецкого.
Дмитриев Александр Борисович, к.м.н., заведующий нейрохирургическим отделением №5 ФГБУ "Федеральный центр нейрохирургии"Министерства здравоохранения Российской Федерации.
Ермоленко Наталия Александровна, д.м.н., зав. кафедрой неврологии ФГОУ ВГМУ им. Н.Н. Бурденко, главный детский невролог департамента здравоохранения Воронежской области, заведующая неврологическим отделением БУЗ ВО «Воронежская областная детская клиническая больница №1».
Жидкова Ирина Александровна, д.м.н., профессор кафедры нервных болезней ФГБОУ ВО МГМСУ им. А.И.Евдокимова Министерства здравоохранения Российской Федерации, член президиума Всероссийского общества неврологов.
Журавлев Дмитрий Викторович, к.м.н., научный сотрудник ГБУЗ «Научно-практический психоневрологический центр имени З.П. Соловьева» Департамента здравоохранения города Москвы.
Заваденко Николай Николаевич, д.м.н., профессор, заведующий кафедрой неврологии, нейрохирургии медицинской генетики имени академика Л.О. Бадаляна педиатрического факультета ФГАОУ ВО «Российский национальный исследовательский медицинский университет имени Н.И. Пирогова» Министерства здравоохранения Российской Федерации.
Зинчук Михаил Сергеевич, к.м.н., ведущий научный сотрудник ГБУЗ «Научно-практический психоневрологический центр имени З.П. Соловьева» Департамента здравоохранения города Москвы.
Иванова Галина Евгеньевна, д.м.н., профессор, главный специалист по медицинской реабилитации Министерства здравоохранения Российской Федерацией, заведующая кафедрой медицинской реабилитации ФДПО ФГАОУ ВО «Российский национальный исследовательский медицинский университет имени Н.И. Пирогова» Министерства здравоохранения Российской Федерации, председатель президиума Общероссийской общественной организации содействия развитию медицинской реабилитологии «Союз реабилитологов России», руководитель НИЦ медицинской реабилитации ФГУ ФЦМН ФМБА России
Каймовский Игорь Леопольдович, к.м.н., заведующий Межокружным отделением пароксизмальных состояний ГКБ им В.М.Буянова, ведущий научный сотрудник ГБУЗ «Научно-практический психоневрологический центр имени З.П. Соловьева» Департамента здравоохранения города Москвы.
Карлов Владимир Алексеевич, д.м.н., член-корреспондент РАН, заслуженный деятель науки РФ, профессор кафедры нервных болезней ФГБОУ ВО МГМСУ им. А.И.Евдокимова Министерства здравоохранения Российской Федерации, президент Российской противоэпилептической лиги.
Ковалева Ирина Юрьевна, к.м.н., профессор кафедры неврологии, нейрохирургии и медицинской генетики лечебного факультета ФГАОУ ВО «Российский национальный исследовательский медицинский университет имени Н.И. Пирогова» Министерства здравоохранения Российской Федерации.
Комольцев Илья Геральдович, невролог, научный сотрудник ГБУЗ «Научно-практический психоневрологический центр имени З.П. Соловьева» Департамента здравоохранения города Москвы.
Крылов Владимир Викторович, д.м.н., академик РАН, главный внештатный специалист по нейрохирургии Министерства здравоохранения Российской Федерации, заведующий кафедрой нейрохирургии и нейрореанимации Московского государственного медико-стоматологического университета имени А.И. Евдокимова.
Кустов Георгий Владимирович, психиатр, научный сотрудник ГБУЗ «Научно-практический психоневрологический центр имени З.П. Соловьева» Департамента здравоохранения города Москвы.
Куцев Сергей Иванович – д.м.н., член-корреспондент РАН, директор ФГБНУ Медико-генетический научный центр имени академика Н.П. Бочкова, заведующий кафедрой молекулярной и клеточной генетики ФГБОУ ВО «Российский национальный исследовательский медицинский университет имени Н.И.Пирогова» Министерства здравоохранения Российской Федерации, главный внештатный специалист по медицинской генетике Минздрава России, председатель Этического Комитета Минздрава России, президент Ассоциации медицинских генетиков России.
Лебедева Анна Валерьяновна, д.м.н., профессор кафедры неврологии, нейрохирургии и медицинской генетики ФГАОУ ВО «Российский национальный исследовательский медицинский университет имени Н.И. Пирогова» Министерства здравоохранения Российской Федерации, член президиума Всероссийского общества неврологов.
Литвиненко Игорь Вячеславович, д.м.н., профессор, главный невролог Министерства обороны Российской Федерации, начальник кафедры и клиники нервных болезней им. М.И. Аствацатурова ФГБВОУ ВО «Военно-медицинская академия им. С. М. Кирова» Министерства обороны Российской Федерации.
Мартынов Михаил Юрьевич, д.м.н., член-корреспондент РАН, главный внештатный специалист по неврологии Министерства здравоохранения Российской Федерации, профессор кафедры неврологии, нейрохирургии и медицинской генетики ФГАОУ ВО «Российский национальный исследовательский медицинский университет имени Н.И. Пирогова» Министерства здравоохранения Российской Федерации, учёный секретарь Всероссийского общества неврологов, член правления Национальной ассоциации по борьбе с инсультом.
Мельникова Елена Валентиновна, д.м.н., заместитель главного врача - руководитель регионального сосудистого центра Санкт-Петербургского государственного бюджетного учреждения здравоохранения "Городская больница № 26", профессор кафедры физических методов лечения и спортивной медицины факультета послевузовского образования федерального государственного бюджетного образовательного учреждения высшего образования «Первый Санкт-Петербургский государственный медицинский университет имени академика И.П. Павлова», заведующая кафедрой клинической медицины и медицинской реабилитации Частного образовательного учреждения высшего образования «Санкт-Петербургский медико-социальный институт», главный внештатный специалист СЗФО РФ по медицинской реабилитации член Президиума общероссийской общественной организации содействия развитию медицинской реабилитологии «Союз реабилитологов России»
Михайлов Владимир Алексеевич, д.м.н., профессор, главный научный сотрудник, научный руководитель отделения интегративной терапии больных психоневрологического профиля и отделения экзогенно-органических расстройств и эпилепсии ФГБУ Национального медицинского исследовательского центра психиатрии и неврологии им. В.М. Бехтерева Министерства здравоохранения Российской Федерации.
Мишина Ирина Алексеевна, к.м.н., старший научный сотрудник Научно-консультативного отделения ФГБНУ «Медико-генетический научный центр имени академика Н.П. Бочкова».
Можейко Елена Юрьевна, д.м.н., профессор, главный специалист по медицинской реабилитации Красноярска, зав. кафедрой медицинской реабилитации Крас ГМУ.
Мухин Константин Юрьевич, д.м.н., профессор, руководитель Института Детской Неврологии и Эпилепсии им. Свт. Луки и Института Детской и Взрослой Неврологии и Эпилепсии им. Свт. Луки.
Невзорова Диана Владимировна, к.м.н., главный врач ГКУЗ «Хоспис № 1 им. В.В. Миллионщиковой» Департамента здравоохранения города Москвы, главный внештатный специалист по паллиативной помощи Минздрава России.
Одинак Мирослав Михайлович, д.м.н., член-корреспондент РАН, профессор кафедры и клиники нервных болезней им. М.И. Аствацатурова ФГБВОУ ВО «Военно-медицинская академия им. С. М. Кирова» Министерства обороны Российской Федерации.
Охрим Инна Владимировна, к.м.н, доцент кафедры неврологии, нейрохирургии и медицинской генетики ФГБОУ ВО Санкт-Петербургский государственный педиатрический медицинский университет Министерства здравоохранения Российской Федерации.
Павлов Николай Александрович, к.м.н., ведущий научный сотрудник кафедры неврологии, нейрохирургии и медицинской генетики ФГАОУ ВО «Российский национальный исследовательский медицинский университет имени Н.И. Пирогова» Министерства здравоохранения Российской Федерации, член президиума Всероссийского общества неврологов.
Пашнин Евгений Вячеславович, психиатр, научный сотрудник ГБУЗ «Научно-практический психоневрологический центр имени З.П. Соловьева» Департамента здравоохранения города Москвы.
Петрухин Андрей Сергеевич, д.м.н., профессор кафедры неврологии, нейрохирургии и медицинской генетики лечебного факультета ФГАОУ ВО «Российский национальный исследовательский медицинский университет имени Н.И. Пирогова» Министерства здравоохранения Российской Федерации, член Президиума Всероссийского общества неврологов, президент Объединения врачей-эпилептологов и пациентов.
Полевиченко Елена Владимировна, д.м.н., профессор кафедры онкологии, гематологии и лучевой терапии ФГАОУ ВО «Российский национальный исследовательский медицинский университет имени Н.И. Пирогова» Министерства здравоохранения Российской Федерации, главный внештатный детский специалист по паллиативной помощи Минздрава России.
Прокопенко Семен Владимирович, д.м.н., профессор, заведующий кафедрой нервных болезней КрасГМУ, главный нейрореабилитолог СФО, научный руководитель службы неврологии и нейрореабилитации ФГБУ ФСНКЦ ФМБА России.
Прокудин Михаил Юрьевич, к.м.н., преподаватель кафедры и клиники нервных болезней им. М.И. Аствацатурова ФГБВОУ ВО «Военно-медицинская академия им. С. М. Кирова» Министерства обороны Российской Федерации.
Рачин Андрей Петрович, д.м.н., профессор, заместитель директора по научной работе, зав. отделом нейрореабилитации и клинической психологии ФГБУ «Национальный медицинский исследовательский центр реабилитации и курортологии» Министерства здравоохранения Российской Федерации.
Ридер Флора Кирилловна, к.м.н., ведущий научный сотрудник ГБУЗ «Научно-практический психоневрологический центр имени З.П. Соловьева» Департамента здравоохранения города Москвы.
Рудакова Ирина Геннадьевна, д.м.н., профессор кафедры неврологии ГБУЗ МО "Московский областной научно-исследовательский клинический институт им. М. Ф. Владимирского", заслуженный работник здравоохранения МО.
Сивакова Наталия Александровна - кандидат медицинских наук, старший научный сотрудник отделения лечения больных с экзогенно-органическими расстройствами и эпилепсией ФГБУ Национальный медицинский исследовательский центр психиатрии и неврологии имени В.М. Бехтерева Министерства здравоохранения Российской Федерации.
Синкин Михаил Владимирович, к.м.н., старший научный сотрудник, руководитель группы нейрофизиологии НИИ скорой помощи им. Н.В.Склифосовского, заведующий лабораторией инвазивных нейроинтерфейсов ФГБОУ ВО МГМСУ им. А.И.Евдокимова Министерства здравоохранения Российской Федерации.
Супонева Наталья Александровна, д.м.н.,профессор РАН,член-корреспондент РАН, заведующая отделением нейрореабилитации и физиотерапии ФГБНУ НЦН РАН РФ.
Талыпов Александр Эрнестович, врач-нейрохирург, д.м.н., ведущий научный сотрудник НИИ скорой помощи им. Н.В.Склифосовского.
Трифонов Игорь Сергеевич, к.м.н., врач-нейрохирург Университетской клиники ФГБОУ ВО МГМСУ им. А.И.Евдокимова Министерства здравоохранения Российской Федерации, доцент кафедры нейрохирургии и нейрореанимации ФГБОУ ВО МГМСУ им. А.И.Евдокимова Министерства здравоохранения Российской Федерации.
Федермессер Анна Константиновна, руководитель ГБУЗ «Московский многопрофильный центр паллиативной помощи ДЗМ».
Фесюн Анатолий Дмитриевич, д.м.н., профессор, директор ФГБУ «Национальный медицинский исследовательский центр реабилитации и курортологии» Министерства здравоохранения Российской Федерации.
Хатькова Светлана Евгеньевна, д.м.н., профессор, главный внештатный специалист-невролог, заведующая отделением медицинской реабилитации взрослых с нарушениями функции центральной и периферической нервной системы НМИЦ ЛРЦ МЗ РФ.
Холин Алексей Александрович, д.м.н., профессор кафедры неврологии, нейрохирургии и медицинской генетики имени академика Бадаляна Л.О. педиатрического факультета ФГАОУ ВО «Российский национальный исследовательский медицинский университет имени Н.И. Пирогова» Министерства здравоохранения Российской Федерации.
Шарков Артем Алексеевич, врач-невролог, научный сотрудник «Научно-исследовательского клинического института педиатрии им. акад. Ю.Е. Вельтищева» ФГАОУ ВО «Российский национальный исследовательский медицинский университет имени Н.И. Пирогова» Министерства здравоохранения Российской Федерации.
Щедеркина Инна Олеговна, к.м.н., руководитель Центра по лечению цереброваскулярной патологии у детей и подростков ГБУЗ «Московская ДГКБ ДЗМ».
Якунина Ольга Николаевна – кандидат психологических наук, старший научный сотрудник отделения лечения больных с экзогенно-органическими расстройствами и эпилепсией ФГБУ Национальный медицинский исследовательский центр психиатрии и неврологии имени В.М. Бехтерева Министерства здравоохранения Российской Федерации.
Все члены рабочей группы заявили об отсутствии финансовой или нематериальной заинтересованности по теме разработанных клинических рекомендаций. Член(ы) рабочей группы, сообщившие об обстоятельства, которые могут повлечь за собой конфликт интересов, был(и) исключен(ы) из обсуждения разделов, связанных с областью конфликта интересов.
Приложение А2. Методология разработки клинических рекомендаций
Приложение А2. Методология разработки клинических рекомендаций
Целевая аудитория данных клинических рекомендаций:
1. Врачи: неврологи, педиатры, терапевты, психиатры, генетики, нейрохирурги, анестезиологи-реаниматологи, врачи функциональной диагностики, лучевой диагностики.
2. Студенты медицинских ВУЗов, ординаторы, аспиранты.
Таблица П1. Шкала оценки уровней достоверности доказательств (УДД) для методов диагностики (диагностических вмешательств)
УДД | Расшифровка |
|---|---|
1 | Систематические обзоры исследований с контролем референсным методом или систематический обзор рандомизированных клинических исследований с применением мета-анализа |
2 | Отдельные исследования с контролем референсным методом или отдельные рандомизированные клинические исследования и систематические обзоры исследований любого дизайна, за исключением рандомизированных клинических исследований, с применением мета-анализа |
3 | Исследования без последовательного контроля референсным методом или исследования с референсным методом, не являющимся независимым от исследуемого метода или нерандомизированные сравнительные исследования, в том числе когортные исследования |
4 | Несравнительные исследования, описание клинического случая |
5 | Имеется лишь обоснование механизма действия или мнение экспертов |
Таблица П2. Шкала оценки уровней достоверности доказательств (УДД) для методов профилактики, лечения и реабилитации (профилактических, лечебных, реабилитационных вмешательств)
УДД | Расшифровка |
|---|---|
1 | Систематический обзор РКИ с применением мета-анализа |
2 | Отдельные РКИ и систематические обзоры исследований любого дизайна, за исключением РКИ, с применением мета-анализа |
3 | Нерандомизированные сравнительные исследования, в т.ч. когортные исследования |
4 | Несравнительные исследования, описание клинического случая или серии случаев, исследования «случай-контроль» |
5 | Имеется лишь обоснование механизма действия вмешательства (доклинические исследования) или мнение экспертов |
Таблица П 3. Шкала оценки уровней убедительности рекомендаций (УУР) для методов профилактики, диагностики, лечения и реабилитации (профилактических, диагностических, лечебных, реабилитационных вмешательств)
УУР | Расшифровка |
|---|---|
A | Сильная рекомендация (все рассматриваемые критерии эффективности (исходы) являются важными, все исследования имеют высокое или удовлетворительное методологическое качество, их выводы по интересующим исходам являются согласованными) |
B | Условная рекомендация (не все рассматриваемые критерии эффективности (исходы) являются важными, не все исследования имеют высокое или удовлетворительное методологическое качество и/или их выводы по интересующим исходам не являются согласованными) |
C | Слабая рекомендация (отсутствие доказательств надлежащего качества (все рассматриваемые критерии эффективности (исходы) являются неважными, все исследования имеют низкое методологическое качество и их выводы по интересующим исходам не являются согласованными) |
Порядок обновления клинических рекомендаций
Механизм обновления клинических рекомендаций предусматривает их систематическую актуализацию – не реже чем один раз в три года или при появлении новой информации о тактике ведения пациентов с данным заболеванием. Решение об обновлении принимает МЗ РФ на основе предложений, представленных медицинскими некоммерческими профессиональными организациями. Сформированные предложения должны учитывать результаты комплексной оценки лекарственных препаратов, медицинских изделий, а также результаты клинической апробации.
Приложение А3. Справочные материалы, включая соответствие показаний к применению и противопоказаний, способов применения и доз лекарственных препаратов, инструкции по применению лекарственного препарата
1. Основные нормативно-правовые акты, регулирующие оказание паллиативной медицинской помощи:
· Федеральный закон "О внесении изменений в Федеральный закон "Об основах охраны здоровья граждан в Российской Федерации" по вопросам оказания паллиативной медицинской помощи" от 06.03.2019 № 18-ФЗ.
· Приказ Минздрава России № 345н, Минтруда России от 31.05.2019 № 372н «Об утверждении положения об организации оказания паллиативной медицинской помощи, включая порядок взаимодействия медицинских организаций, организаций социального обслуживания и общественных объединений, иных некоммерческих организаций, осуществляющих свою деятельность в сфере охраны здоровья».
· Приказ Минздрава России № 348н от 31 мая 2019 года «Об утверждении перечня медицинских изделий, предназначенных для поддержания органов и систем организма человека, предоставляемых для использования на дому».
· Приказ Минздрава России № 505н от 10 июля 2019 года «Об утверждении Порядка передачи от медицинской организации пациенту (его законному представителю) медицинских изделий, предназначенных для поддержания функций органов и систем организма человека, для использования на дому при оказании паллиативной медицинской помощи».
· Приказ МЗ РФ № 831 от 3 октября 2019 года «Об утверждении ведомственной целевой программы «Развитие системы оказания паллиативной медицинской помощи».
2. Инструкция по применению мидазолама.
3. Инструкция по применению диазепама.
4. Основные механизмы действия и способы применения противоэпилептических препаратов.
Следует учитывать, что у новых и новейших противоэпилептических препаратов инструкции к применению могут обновляться по показаниям, побочным эффектам, дозам и возрастным периодам.
Бензобарбитал **
Механизм действия. Усиливает тормозные ГАМК-ергические влияния в центральной нервной системе, особенно в таламусе, восходящей активирующей ретикулярной формации ствола головного мозга на уровне вставочных нейронов. Блокада натриевых каналов мембраны нервных волокон.
Способ применение и дозы.
Рекомендован детям старше 6 лет и взрослым.
Детям 7 - 10 лет - по 50 - 100 мг на прием (150 - 300 мг/сут), 11 - 14 лет - по 100 мг на прием (300 - 400 мг/сут). Максимальные дозы для детей (старшего возраста): разовая - 150 мг, суточная - 450 мг.
Взрослым: перорально начиная с 100 мг в вечернее время. Увеличение дозы возможно через 3 дня на 100 мг до достижения клинического эффекта. Средняя суточная доза составляет — 300 - 600 мг в сутки в 3 приема.
Бриварацетам**
Рекомендован подросткам с 16 лет и взрослым пациентам.
Механизм действия. Обратимый и селективный лиганд для синаптических пузырьков 2А (SV2A) в головном мозге. Хотя точная роль данного белка неизвестна, было показано, что он модулирует экзоцитоз нейротрансмиттеров. Имеет в 25 раз более высокую аффинность, чем левитерацетам.
Способ применение и дозы. Внутрь, не разжевывая, запивая водой, независимо от приема пищи. Рекомендуемая начальная доза составляет 50 мг/сутки или 100 мг/сутки по решению лечащего врача, исходя из требуемого противосудорожного эффекта и потенциального побочного действия. Суточная доза делится поровну на два приема утром и вечером. В зависимости от индивидуального ответа и переносимости, доза может быть изменена в пределах от 50 мг/сутки до 200 мг/сутки.
Вальпроевая кислота**
Механизм действия. Вальпроевая кислота** ингибирует вольтаж-зависимые натриевые каналы, блокирует кальциевые каналы Т-типа, а также воздействует на ГАМК-ергическую систему. Полагают, что вальпроевая кислота** ингибирует ГАМК-трансаминазу (фермент, расщепляющий ГАМК) и повышает активность декарбоксилазы глутаминовой кислоты (фермент, участвующий в синтезе ГАМК). Вальпроевая кислота увеличивает вызванное ГАМК постсинаптическое ингибирование.
Способ применение и дозы.
Способ применения и режим дозирования конкретного препарата зависят от его формы выпуска и других факторов. Пролонгированные формы применяются 2 раза в сутки; непролонгированные формы – 3 раза в сутки.
У детей, которые не могут глотать таблетки, целесообразно применение специальных лекарственных форм вальпроевой кислоты** (капель, сиропа, микрогранул). У пациентов любого возраста целесообразен прием вальпроевой кислоты** в виде лекарственных форм пролонгированного действия, что улучшает переносимость и дает дополнительный противосудорожный эффект.
Для приема внутрь у детей с массой тела более 25 кг начальная доза составляет 10 - 15 мг/кг/сут. Затем дозу постепенно увеличивают на 200 мг/сут с интервалом 3 - 4 дня до достижения клинического эффекта. Средняя суточная доза составляет 20 - 30 мг/кг.
Для детей с массой тела менее 25 кг и новорожденных средняя суточная доза составляет 20 - 30 мг/кг.
Для взрослых: внутрь начиная с 300 - 500 мг/сутки (в два приема) с постепенным увеличением на 250 - 300 мг/неделю до достижения поддерживающей дозы 600 - 3000 мг/сутки. Терапевтическая концентрация в плазме крови — 50 - 150 мкг/мл.
Вальпроевая кислота** ингибирует микросомальные ферменты (цитохромы) печени, поэтому ее применение вызывает увеличение концентрации в крови других ПЭП (фенобарбитала**, ламотриджина, карбамазепина**).
Габапентин
Механизм действия. Ингибирует потенциал-зависимые кальциевые каналы связываясь с альфа-2-дельта субъединицей данного канала, вызывая снижение выброса нейротрансмиттера.
Способ применение и дозы.
Для взрослых и детей с 12 лет: терапию можно начинать с дозы по 300 мг 3 раза в сутки в 1-й день или увеличивать постепенно до 900 мг/сут по схеме: 300 мг 1 раз в сут. – 1 день, 300 мг х 2 раза в сут. – 2-й день, далее 300 мг х 3р в сут. Эффективная среднетерапевтическая доза 900 –2400 мг/сут. Максимальная доза 3600 мг/сут.
Диазепам
Механизм действия. Относится к группе производных бензодиазепина. Механизм действия диазепама тесно связан с тормозным эндогенным нейромедиатором гамма-аминомасляной кислотой (ГАМК) и рецептором ГАМКА. Диазепам усиливает тормозное влияние ГАМК-ергических нейронов в ЦНС, стимулирует бензодиазепиновые рецепторы, усиливает пресинаптическое торможение.
Диазепам (раствор ректальный) предназначен для лечения продолжительных (более 2 - 3 минут) острых судорожных приступов при эпилепсии у младенцев с 6 месяцев, детей грудного, дошкольного, младшего школьного возраста и подростков (до 18 лет).
Способ применения и дозы.
Ректально. Содержимое одной микроклизмы должно быть введено полностью за одно применение. Доза диазепама подбирается индивидуально в зависимости от состояния пациента, его возраста, массы тела, вида и тяжести заболевания. Детям с массой тела менее 15 кг назначают 5 мг; детям с массой тела более 15 кг – 10 мг. Максимальный эффект развивается через 11 - 23 минуты. При введении высоких доз необходим тщательный медицинский контроль и мониторинг состояния пациента. Если приступ не купировался в течение 10 минут после применения диазепама, необходимо обратиться за экстренной медицинской помощью и передать пустой тюбик врачу, чтобы предоставить ему информацию о дозе, полученной пациентом.
Диазепам ** (раствор для внутривенного и внутримышечного введения).
Взрослым при эпилептическом статусе назначают в/м или в/в по 10-20 мг. При необходимости дозу повторяют через 3 - 4 часа.
Детям назначают после 5-й недели жизни в/в медленно по 0,1 – 0,3 мг/кг массы тела до максимальной дозы 5 мг. При необходимости инъекцию повторяют через 2-4 часа в зависимости от клинической ситуации.
Детям от 5 лет и старше по 1 мг в/в медленно каждые 2 - 5 мин до максимальной дозы 10 мг. При необходимости лечение можно повторить через 2 - 4 часа.
Зонисамид
Механизм действия. Блокатор потенциал-зависимых натриевых каналов, ингибитор кальциевых каналов Т-типа, блокирует карбоангидразу.
Показания: Монотерапия у взрослых пациентов с парциальными эпилептическими приступами с вторичной генерализацией или без, с впервые диагностированной эпилепсией. В составе дополнительной терапии у взрослых, подростков и детей с 6 лет с парциальными эпилептическими приступами с вторичной генерализацией или без.
Способ применения и дозы.
Внутрь, запивая водой, вне зависимости от приема пищи. Доза препарата подбирается с учетом лечебного эффекта.
Взрослые пациенты. Эффективной является суточная доза 300-500 мг. Для монотерапии: начальная доза 100 мг/сут однократно. Увеличение на 100 мг с двухнедельным интервалом до максимальной рекомендованной дозы 500 мг. Для дополнительной терапии: начальная доза - 50 мг/сут, разделенные на два приема. На 2 - 3-й неделе можно увеличить дозу до 100 мг/сут. Далее увеличение не более, чем на 100 мг каждые 7 дней (для пациентов, принимающих индукторы ферментов печени) и на 100 мг каждые 2 недели (для пациентов, не принимающих индукторы ферментов печени, пациентов с почечной или печеночной недостаточностью) до максимальной рекомендованной дозы 500 мг в день.
Подростки и дети с 6 лет. Начальная доза 1 мг/кг в сутки (однократно) с увеличением дозы на 1мг/кг с недельными интервалами при сопутствующем приеме индукторов ферментов печени. И с шагом 1 мг/кг c двухнедельными интервалами при отсутствии приема индукторов ферментов печени. Доза препарата подбирается с учетом клинического эффекта.
Карбамазепин **
Механизм действия. Блокатор потенциалзависимых натриевых каналов.
Способ применение и дозы.
Рекомендован к применению для взрослых и детей от 3 лет в виде таблеток.
Для взрослых стартовая доза составляет 100 - 200 мг/сут внутрь в 1 - 2 приема с постепенным увеличением на 200 мг/неделю до достижения поддерживающей дозы 600 - 1200 мг/сутки. Максимальная доза — 1600 мг/сутки. Терапевтическая концентрация в плазме крови — 4 - 12 мкг/мл. Частота приема — 3 раза в сутки. Целесообразно применение ретардных форм 1 - 2 раза в сутки.
Детям старше 3-х лет – начальная доза 100 мг/сут, с постепенным повышением на 100 мг каждую неделю. Поддерживающие дозы 10 - 20 мг/кг в сут (в несколько приемов).
Детям 4 - 5 лет – 200 - 400 мг/сут. в 1-2 приема.
Детям 6 - 10 лет – 400 - 600 мг/сут в 2 - 3 приема.
Детям 11 - 15 лет – 600 - 1000 мг/сут в 2 - 3 приема.
Подросткам старше 15 лет – 800 – 1200 мг/сут в 2 - 3 приема (как для взрослых).
Клоназепам **
Механизм действия. Усиливает ингибирующее действие ГАМК.
Способ применение и дозы.
Рекомендован к применению для взрослых и детей от 3 лет в виде таблеток.
Взрослые.
Начальная доза должна быть не более 1,5 мг/сут., разделенная на 3 приема (0,5 мг 3 раза в день) внутрь. Дозу необходимо постепенно увеличивать на 0,5 – 1 мг через каждые 3 дня. Поддерживающая доза устанавливается индивидуально для каждого пациента в зависимости от клинического эффекта (4 - 8 мг/сут в 3 - 4 приема). Максимальная суточная доза — 20 мг/сут.
Дети 3 - 5 лет.
Начальная доза 0,25 мг/сут. Поддерживающая доза 1 - 3 мг/сут.
Дети 6 - 12 лет.
Начальная доза 0,5 мг/сут. Поддерживающая доза 3 - 6 мг/сут. Максимальная суточная доза для детей составляет 0,2 мг/кг/сут.
Пациенты пожилого возраста.
Начальная доза должна быть не более 0,5 мг/сут.
Клобазам.
Механизм действия.
Производное бензодиазепина. Усиливает ингибирующее действие ГАМК.
Рекомендован в качестве дополнительной терапии для пациентов с эпилепсией, не достигших ремиссии на терапии одним или более ПЭП.
Способ применения и дозы.
Внутрь в виде таблеток для взрослых и детей старше 3-х лет, вне зависимости от приема пищи, целиком, либо измельчив и смешав с яблочным пюре. Таблетка 10 мг может быть разделена на равные половинки по 5 мг каждая. При лечении эпилепсии рекомендуемая начальная доза составляет 20 - 30 мг/сут. При необходимости дозу можно увеличить до 60 мг/сут.
Детский возраст до 6 лет.
У детей лечение следует начинать с минимальной дозы 5 мг/сут; поддерживающая доза составляет 0,3 - 1 мг/кг массы тела в сутки. На сегодняшний день не существует лекарственной формы препарата для безопасного и точного его дозирования при лечении детей младше 6 лет, в связи с чем невозможно дать рекомендации в отношении дозы препарата, которую можно применять у детей данной возрастной категории.
Лакосамид**
Лекарственные формы: таблетки, раствор для приема внутрь, раствор для инфузий.
Механизм действия. Избирательно усиливает медленную инактивацию потенциалзависимых натриевых каналов, что ведет к стабилизации гипервозбудимых мембран нейронов.
Способ применение и дозы.
Таблетки, покрытые пленочной оболочкой для приема внутрь (50 мг, 100 мг, 150 мг, 200 мг). Рекомендованы для взрослых и подростков с эпилепсией с 16 лет.
Внутрь в 2 приема — утром и вечером, вне зависимости от времени приема пищи. Стартовая доза составляет 50 мг 2 раза в день. Через 1 нед дозу увеличивают до 100 мг 2 раза в день. С учетом эффективности и переносимости поддерживающую дозу можно увеличить до 150 мг 2 раза в день на 3-й неделе, до 400 мг/день (200 мг 2 раза в день) с 4-й недели. Максимальная рекомендуемая доза 600 мг/с (для монотерапии).
Раствор для приема внутрь.
Рекомендован для приема внутрь взрослым, подросткам и детям 4 лет и старше как в монотерапии, так и в дополнительной терапии эпилепсии. Суточную дозу делят на 2 приема – обычно утром и вечером, вне зависимости от приема пищи.
Подростки и дети с массой тела 50 кг и выше, а также взрослые.
Монотерапия. Рекомендуемая начальная доза составляет 50 мг 2 раза в день с последующим увеличением до начальной терапевтической дозы 100 мг 2 раза в день по истечении первой недели лечения. В зависимости от ответа и переносимости, поддерживающая доза может быть увеличена на 100 мг/сут (50 мг х 2р в день) с интервалами в неделю. Максимальная суточная поддерживающая доза 600 мг/сут (300 мг х 2р в день).
Дополнительная терапия. Рекомендуемая начальная доза составляет 50 мг 2 раза в день с последующим увеличением до начальной терапевтической дозы 100 мг 2 раза в день по истечении первой недели лечения. В зависимости от ответа и переносимости, доза может быть увеличена на 100 мг/сут (50 мг х 2р в день) с интервалами в неделю. Рекомендуемая максимальная суточная поддерживающая доза 400 мг/сут (200 мг х 2р в день).
Дети (старше 4 лет) и подростки с массой тела до 50 кг. Доза определяется на основании массы тела. Поэтому рекомендуется начинать лечение с раствора для приема внутрь, а затем переходить на таблетки (по желанию). При назначении раствора для приема внутрь дозу следует выражать в объемных (мл), а не в весовых единицах (мг).
Монотерапия. Рекомендуемая начальная доза составляет 2 мг/кг/сут, которая должна быть увеличена до начальной терапевтической дозы 4 мг/кг/сут по истечении первой недели. В зависимости от ответа и переносимости лечения, поддерживающая доза может быть увеличена еще на 2 мг/кг/сут с интервалами в неделю. Дозу следует повышать постепенно до получения оптимального ответа. У детей с массой тела до 40 кг максимальная рекомендованная доза составляет 12 мг/кг/сут. У детей с массой тела от 40 кг до 50 кг максимальная рекомендованная доза составляет 10 мг/кг/сут.
Раствор для инфузий назначают взрослым пациентам и подросткам с 16 лет с эпилепсией в тех случаях, когда временно невозможна пероральная терапия. Общая продолжительность лечения внутривенной формой лакосамида находится на усмотрении врача.
Ламотриджин.
Механизм действия. Воздействует на вольтаж-зависимые натриевые каналы и блокирует выброс нейротрансмиттеров, в первую очередь глутамата.
Способ применение и дозы.
При приеме внутрь для взрослых и детей старше 12 лет начальная доза при монотерапии составляет 25 мг один раз в день в течение 2 недель; в последующие 2 недели – по 50 мг один раз в день. В дальнейшем, каждые одну-две недели можно повышать суточную дозу на 50 мг до тех пор, пока не будет достигнут оптимальный эффект. Обычно суточная поддерживающая доза составляет 100 - 200 мг. При этом положительный эффект может быть достигнут в некоторых случаях и на больших дозировках: 400 - 500 мг/сутки.
У пациентов, принимающих вальпроевую кислоту**, начальная суточная доза ламотриджина в течение двух недель должна составлять 25 мг через день; в течение следующих двух недель ежедневно принимать по 25 мг один раз в день. В последующем, каждые 1 - 2 недели дозу можно увеличивать на 25 - 50 мг, до тех пор, пока не будет достигнут оптимальный эффект. Обычно поддерживающая суточная доза составляет 100 - 200 мг.
У взрослых, принимающих ПЭП, индуцирующие ферменты печени, лечение начинают с 50 мг в течение двух недель. В течение последующих двух недель 100 мг/сутки распределенные на два приема. В дальнейшем, каждые 1-2 недели можно повышать дозу не более, чем на 100 мг до получения оптимального эффекта.
Дети в возрасте от 3 до 12 лет.
Монотерапия. Начальная доза препарата при монотерапии пациентов с типичными абсансами составляет 0,3 мг/кг/сут в 1 или 2 приема в течение 2-х недель с последующим повышением дозы до 0,6 мг/кг/сут в 1 или 2 приема в течение следующих 2 недель. Затем дозу препарата следует увеличивать максимально на 0,6 мг/кг каждые 1 - 2 недели до достижения оптимального терапевтического эффекта. Обычная поддерживающая доза составляет от 1 до 10 мг/кг/сут в 1 или 2 приема, хотя некоторым пациентам с типичными абсансами требуются более высокие дозы.
Комбинированная терапия. У пациентов, принимающих вальпроевую кислоту** в сочетании с другими ПЭП или без них, начальная доза препарата составляет 0,15 мг/кг
сут 1 раз в день в течение 2 недель; в дальнейшем – 0,3 мг/кг/сут – 1 раз в сутки в течение 2 недель. Затем доза может быть увеличена максимально на 0,3 мг/кг каждые 1 - 2 недели до достижения оптимального терапевтического эффекта. Обычная поддерживающая доза составляет 1 - 5 мг/кг/сут в 1 или 2 приема. Максимальная доза составляет 200 мг/сут.
У пациентов, которые получают ПЭП или другие препараты, индуцирующие глюкуронизацию ламотриджина, в сочетании с другими ПЭП или без них (за исключением вальпроевой кислоты **), начальная доза препарата составляет 0,6 мг/кг/сут в 2 приема в течение 2 недель. Затем дозу следует увеличивать максимально на 1,2 мг/кг каждые 1 - 2 недели до достижения оптимального терапевтического эффекта. Обычная поддерживающая доза составляет 5 - 15 мг/кг/сут в 2 приема. Максимальная доза составляет 400 мг/сут.
Леветирацетам**
Механизм действия. Связывается с белком синаптических пузырьков 2А (synapticvesicle 2A, SV2A). Опосредованная модуляция выделения нейротрансмиттера через модификацию белка синаптических везикул (SV2A).
Способ применение и дозы.
Таблетки покрытые пленочной оболочкой.
Внутрь, независимо от приема пищи. Суточную дозу препарата делят на два приема в одинаковой дозе.
Монотерапия. Взрослым и подросткам с 16 лет лечение следует начинать с суточной дозы 500 мг, разделенной на 2 приема (по 250 мг 2 раза в сутки). Через 2 недели доза может быть увеличена до начальной терапевтической – 1000 мг (по 500 мг 2 раза в сутки). Максимальная суточная доза составляет 3000 мг (по 1500 мг 2 раза в сутки).
В составе дополнительной терапии.
Взрослым и подросткам (от 12 до 17 лет) с массой тела более 50 кг лечение следует начинать с суточной дозы 1000 мг, разделенной на 2 приема (по 500 мг 2 раза в сутки). В зависимости от клинического ответа и переносимости суточная доза может быть увеличена до максимальной – 3000 мг (по 1500 мг 2 раза в сутки). Изменение дозы на 500 мг 2 раза в сутки может осуществляться каждые 2 - 4 недели.
Детям с 6 лет и подросткам (от 12 до 17 лет) с массой тела менее 50 кг лечение следует начинать с суточной дозы 20 мг/кг массы тела, разделенной на 2 приема (по 10 мг/кг массы тела 2 раза в сутки). Изменение дозы на 20 мг/кг массы тела (по 10 мг/кг массы тела 2 раза в сутки) может осуществляться каждые 2 недели до достижения рекомендуемой суточной дозы – 60 мг/кг массы тела (по 30 мг/кг массы тела 2 раза в сутки). Врач должен назначить препарат в наиболее подходящей лекарственной форме и дозировке в зависимости от возраста, массы тела пациента и необходимой терапевтической дозы. В связи с отсутствием нужной дозировки таблетки не предназначены для лечения детей весом менее 25 кг, при назначении дозы менее 250 мг, а также для пациентов, имеющих трудности при глотании. В этих случаях рекомендуется начинать лечение с приема препарата в форме раствора для приема внутрь. Детям с массой тела более 50 кг дозирование осуществляют по схеме, приведенной для взрослых.
Раствор для приема внутрь.
Внутрь, независимо от приема пищи. Суточную дозу препарата делят на два приема в одинаковой дозе.
Монотерапия. Взрослым и подросткам с 16 лет лечение следует начинать с суточной дозы 500 мг, разделенной на 2 приема (по 250 мг 2 раза в сутки). Через 2 недели доза может быть увеличена до начальной терапевтической – 1000 мг (по 500 мг 2 раза в сутки). Максимальная суточная доза составляет 3000 мг (по 1500 мг 2 раза в сутки).
В составе дополнительной терапии. Взрослым c 18 лет и подросткам (от 12 до 17 лет) с массой тела более 50 кг лечение следует начинать с суточной дозы 1000 мг, разделенной на 2 приема (по 500 мг 2 раза в сутки). В зависимости от клинической реакции и переносимости препарата суточная доза может быть увеличена до максимальной – 3000 мг (по 1500 мг 2 раза в сутки). Изменение дозы на 500 мг 2 раза в сутки может осуществляться каждые 2 - 4 недели.
Детям в возрасте от 6 месяцев до 23 месяцев, детям в возрасте от 2 лет до 11 лет и подросткам от 12 лет до 17 лет с массой тела менее 50 кг. Лечение следует начинать с дозы 20 мг/кг массы тела, разделенной на 2 приема (по 10 мг/кг массы тела 2 раза в сутки). В зависимости от клинической реакции и переносимости препарата суточная доза может быть увеличена до 30 мг/кг 2 раза в сутки. Изменение дозы на 20 мг/кг (по 10 мг/кг массы тела 2 раза в сутки) массы тела может осуществляться каждые 2 недели. Следует применять минимальную эффективную дозу.
Дети в возрасте от 1 месяца до 6 месяцев. Начальная лечебная доза равна 7 мг/кг два раза в сутки. В зависимости от клинической эффективности и переносимости, доза может быть увеличена до 21 мг/кг два раза в сутки. Изменение дозы не должно превышать плюс или минус 7 мг/кг два раза в сутки каждые две недели. Следует назначать минимальную эффективную дозу.
Мидазолам**.
Механизм действия.
Бензодиазепин короткого действия, производное группы имидобензодиазепинов. Стимулирует в мембранах нейронов бензодиазепиновые рецепторы, повышая чувствительность ГАМКА – рецепторов к ГАМК, усиливая процессы торможения в ЦНС.
Способ применения и дозы.
Мидазолам (раствор защечный) предназначен для лечения продолжительных (более 2 - 3 минут) острых судорожных приступов при эпилепсии у младенцев, детей грудного, дошкольного, младшего школьного возраста и подростков от 3 месяцев до 18 лет.
Лечение младенцев в возрасте 3-6 месяцев должно проводиться в условиях стационара при возможности мониторинга состояния и наличии реанимационного оборудования. Рекомендованные дозы указаны в таблице.
Возрастной диапазон | Доза | Форма выпуска |
|---|---|---|
от 3 до 6 месяцев | 2,5 мг | Тюбик по 1 мл – 2,5 мг мидазолама (2,5 мг/мл) |
от 6 месяцев до 1 года | 2,5 мг | Тюбик по 1 мл – 2,5 мг мидазолама (2,5 мг/мл) |
от 1 года до 5 лет (включит.) | 5,0 мг | Тюбик по 1 мл – 5.0 мг мидазолама (5 мг/мл) |
от 5 лет до 10 лет (включит) | 7,5 мг | Тюбик по 1,5 мл – 7,5 мг мидазолама (5 мг/мл) |
от 10 лет до 18 лет | 10,0 мг | Тюбик по 2 мл – 10,0 мг мидазолама (5 мг/мл) |
Мидазолам (раствор защечный) предназначен для нанесения на слизистую оболочку полости рта. Необходимо в полном объеме медленно ввести раствор в пространство между десной и щекой. При необходимости (при больших объемах и/или у маленьких пациентов) половину дозы вводят медленно за одну щеку, а затем вторую половину – медленно за другую щеку.
#Мидазолам**(раствор для внутривенного или внутримышечного введения 5 мг/мл)
Применяется для лечения эпилептического статуса судорожных приступов у детей и взрослых.
Препарат вводится в/м однократно в дозе 10 мг при массе тела более 40 кг и 5 мг при массе 13 - 40 кг [369].
#Мидазолам** вводится внутримышечно однократно в дозе 0,2 — 0,3 мг/кг. Разовая доза не должна превышать: для детей до 5 лет – 5 мг (1 мл), старше 5 лет - 10 мг (2мл); внутривенно 400 мкг/кг. Возможно внутривенное введение #мидазолама** 0,2 - 0,3 мг/кг болюсно с последующие инфузией 2 - 10 мкг/кг/мин. [536,537].
Окскарбазепин **
Механизм действия. Блокада потенциалзависимых Na+-каналов, а также модуляция К+ и Са+-каналов.
Способ применение и дозы.
Принимают внутрь.
Взрослые. Начальная доза составляет 600 мг/сутки в 2 приема. Дозу при необходимости увеличивают на 600 мг с интервалами в 1 неделю. Средняя терапевтическая доза — 600 - 2400 мг/сутки.
Дети от 1 месяца и подростки.
Начальная доза — 8 - 10 мг/кг массы тела/сут, разделенные на 2 приема. Далее дозу корректируют в зависимости от схемы лечения, возраста пациента, эффективности лечения, функции почек. У детей младше 3 лет препарат следует применять в форме сиропа в связи с трудностями применения твердых лекарственных форм у этой возрастной группы.
Перампанел**
Механизм действия. Селективный неконкурентный антагонист ионотропных АМРА-глутаматных рецепторов на постсинаптических нейронах.
Способ применение и дозы.
Перампанел** принимают внутрь 1 раз в сутки перед сном независимо от приема пищи. Таблетку проглатывают целиком (нельзя делить, разжевывать). Стартовая доза составляет 2 мг/сут. Доза может быть увеличена в зависимости от клинического ответа и переносимости с шагом 2 мг (один раз в неделю либо один раз в две недели). Средняя терапевтическая доза 4 - 8 мг/сутки. Максимальная доза — 12 мг/сутки.
Взрослые и подростки с 12 лет.
Для дополнительного лечения парциальных приступов с вторично-генерализованными приступами или без них с эпилепсией; для дополнительного лечения первично-генерализованных тонико-клонических приступов при идиопатической генерализованной эпилепсии.
Дети с 4 до 11 лет для дополнительного лечения парциальных приступов с вторично-генерализованными приступами или без них с эпилепсией.
Дети в возрасте от 7 до 11 лет для дополнительного лечения первично-генерализованных тонико-клонических приступов при идиопатической генерализованной эпилепсии.
Противопоказание: дети с 4 до 11 лет с массой тела менее 30 кг (для данной лекарственной формы).
Прегабалин**
Механизм действия. Опосредован через дополнительную субъединицу (α2-дельта-протеин) вольтаж-зависимых Са+-каналов в ЦНС.
Способ применение и дозы.
Внутрь, независимо от приема пищи. Препарат применяют в дозе от 150 до 600 мг/сут в 2 или 3 приема.
Рекомендован взрослым пациентам в качестве дополнительной терапии эпилепсии с парциальными судорожными припадками, сопровождающимися или не сопровождающимися вторичной генерализацией.
Начальная доза прегабалина** составляет 150 мг/сут. С учетом достигнутого эффекта и переносимости через 1 неделю дозу можно увеличить до 300 мг/сут, а еще через неделю - до 450 мг/с. Максимальная доза 600 мг/сут.
Примидон
Механизм действия. По химическому строению близок к фенобарбиталу**, но отличается более сильным противосудорожным действием, не оказывая общего угнетающего влияния на центральную нервную систему.
Способ применение и дозы.
Таблетки примидон принимают внутрь после приема пищи.
Дозы подбирают индивидуально, начиная с разовой дозы 125 мг однократно внутрь после еды, затем каждые 3 дня суточную дозу увеличивают на 125 мг до достижения 500 мг/сут. Затем каждые 3 дня дозу повышают на 250 мг для взрослых и детей старше 9 лет; на 125 мг - для детей от 3 до 9 лет, до достижения необходимого эффекта. Максимальная суточная доза для взрослых и детей с 9 лет - 1500 мг/сут; для детей 3 - 9 лет - 1000 мг/сут (в 2 приема).
Руфинамид.
Механизм действия.
Руфинамид модулирует активность натриевых каналов, пролонгируя их неактивное состояние.
Способ применения и дозы.
Внутрь, 2 раза в день (утром и вечером) в равных дозах, запивая водой, во время еды. Если пациенту трудно глотать, таблетки можно измельчить и принять, добавив измельченную таблетку в половину стакана воды.
Применение у детей в возрасте от 1 до 4 лет.
Пациенты, не принимающие вальпроевую кислоту**. Лечение следует начинать с суточной дозы 10 мг/кг, разделенной на 2 приема примерно через каждые 12 часов. В соответствии с клиническим ответом и переносимостью дозу можно повышать в каждый третий день на 10 мг/кг/сутки до целевой дозы 45 мг/кг/сутки, разделенной на 2 приема примерно через каждые 12 часов. Максимальная рекомендованная доза для данной группы пациентов составляет 45 мг/кг/сутки.
Пациенты, принимающие вальпроевую кислоту**. Так как вальпроевая кислота** значительно снижает клиренс руфинамида, то пациентам, принимающим одновременно вальпроевую кислоту**, рекомендуется принимать руфинамид в меньшей максимальной дозе. Лечение следует начинать с суточной дозы 10 мг/кг, разделенной на 2 приема примерно через каждые 12 часов. В соответствии с клиническим ответом и переносимостью дозу можно повышать в каждый третий день на 10 мг/кг/сутки до целевой дозы 30 мг/кг/сутки, разделенной на 2 приема примерно через каждые
12 часов. Максимальная рекомендованная доза для данной группы пациентов составляет 30 мг/кг/сутки.
Применение у детей в возрасте старше 4 лет с массой тела менее 30 кг.
Пациенты с массой тела менее 30 кг, не принимающие вальпроевую кислоту**. Лечение следует начинать с суточной дозы 200 мг. В соответствии с клиническим ответом и переносимостью дозу можно повышать в каждый третий день на 200 мг/сутки до максимальной рекомендованной дозы 1000 мг/сутки.
Пациенты с массой тела менее 30 кг, также принимающие вальпроевую кислоту**. Лечение следует начинать с суточной дозы 200 мг. В соответствии с клиническим ответом и переносимостью дозу можно повышать не ранее чем через 2 дня на 200 мг/сутки до максимальной рекомендованной дозы 600 мг/сутки.
Применение у взрослых, подростков и детей в возрасте старше 4 лет с массой тела более 30 кг. Пациенты с массой тела более 30 кг, не принимающие вальпроевую кислоту**: Лечение следует начинать с суточной дозы 400 мг. В соответствии с клиническим ответом и переносимостью дозу препарата можно повышать через день на 400 мг/сут до максимальной рекомендованной дозы, как указано в таблице ниже.
Масса тела | 30,0- 50,0 кг | 50,1 – 70,0 кг | ≥70,1 кг |
|---|---|---|---|
Максимальная рекомендованная доза | 1800 мг/сутки | 2400 мг/сутки | 3200 мг/сутки |
Пациенты с массой тела более 30 кг, принимающие вальпроевую кислоту**. Лечение следует начинать с суточной дозы 400 мг. В соответствии с клиническим ответом и переносимостью дозу препарата можно повышать через день на 400 мг/сутки до максимальной рекомендованной дозы, как указано в таблице ниже.
Масса тела | 30,0 – 50,0 кг | 50,1 – 70,0 кг | ≥70,1 кг |
|---|---|---|---|
Максимальная рекомендованная доза | 1200 мг/сутки | 1600 мг/сутки | 2200 мг/сутки |
Топирамат**
Рекомендован к применению детям старше 2 лет, подросткам и взрослым пациентам для лечения впервые диагностированной эпилепсии (монотерапия и в составе комплексной терапии), синдроме Леннокса - Гасто (в составе комплексной терапии).
Механизм действия. Модуляция активности вольтаж-зависимых натриевых каналов, потенцирование вызванного ГАМК-потока ионов хлора, ингибирование кальциевых каналов, блокада каинатных рецепторов глутамата, ингибирование изоферментов карбоангидразы.
Способ применение и дозы.
Топирамат** принимают внутрь, вне зависимости от приема пищи. При монотерапии:
Дети: начальная доза — исходя из расчета 0,5 – 1 мг топирамата** на кг веса ребёнка, один раз в сутки, перед сном, в течение недели, далее каждые одну или две недели дозу топирамата** повышают на 0,5 – 1 мг на кг веса в сутки, разделяя её на два приема, до достижения клинического эффекта; рекомендуемая доза — 3 – 6 мг на кг веса ребёнка (в некоторых случаях — до 500 мг в сутки).
Взрослые: начальная доза — 25 мг топирамата один раз в сутки, перед сном, в течение недели, далее каждые одну-две недели дозу топирамата** поднимают на 25 – 50 мг в сутки, разделяя её на два приема, до достижения клинического эффекта; рекомендуемая доза — 100 мг топирамата** в сутки, но не более 500 мг топирамата в сутки (иногда — до 1000 мг в сутки).
В составе комплексной терапии доза топирамата назначается индивидуально, в зависимости от состояния пациента и типа лечения.
Фенитоин**
Механизм действия. В основном связан с влиянием на вольтаж-зависимые натриевые каналы.
Способ применение и дозы.
Для взрослых начальная доза составляет 100 мг (1 таб) от 2 до 4 раз в день. В последующие 7 - 10 дней возможно повышение дозы до максимальной 600 мг/сут. Стандартная поддерживающая доза составляет от 200 до 500 мг/сут, разделенная на несколько приемов.
Детям - 5 мг/кг/сут в два приема с последующим увеличением дозы не более 300 мг/сут. Поддерживающие дозы — 4 - 8 мг/кг/сут.
Фенобарбитал**
Механизм действия. Действие обусловлено активацией ГАМК-ергической системы, влиянием на потенциалзависимые натриевые каналы, а так же подавлением активности глутамата.
Способ применение и дозы.
Внутрь.
Режим дозирования устанавливают строго индивидуально в зависимости от показаний, течения заболевания, переносимости, возраста. Лечение необходимо начинать с наименьшей эффективной дозы.
Дети: 5 - 8 мг/кг в сут (противопоказания: детский возраст до 3 лет для твердой лекарственной формы).
Взрослые: перорально начиная с 50 мг на ночь. Увеличение дозы возможно через 3 дня на 50 мг до достижения клинического эффекта. Средняя суточная доза составляет — 100 - 200 мг в 2 приема.
Эсликарбазепин
Механизм действия. Медленная инактивация потенциал-зависимых Na+-каналов, дополнительный антиэпилептический эффект связан с влиянием на Са2+-каналы.
Способ применение и дозы.
Взрослые пациенты (старше 18 лет).
Принимают внутрь независимо от приема пищи. Таблетку можно делить на две равные части. Рекомендованная начальная доза — 400 мг 1 раз в сутки, через 1 - 2 нед дозу повышают до 800 мг 1 раз в сутки, максимальная доза может быть повышена до 1600 мг при монотерапии и 1200 мг при дополнительной терапии, однократно в сутки.
Этосуксимид**
Механизм действия. Влияние на вольтажзависимые кальциевые каналы (Т-каналы).
Способ применение и дозы.
Капсулы следует проглатывать целиком с достаточным количеством жидкости (например, стакан воды), во время или после еды.
У детей (с 6 лет) и у взрослых лечение начинают с общей суточной дозы, находящейся в диапазоне от 5 до 10 мг/кг массы тела. Общая суточная доза этосуксимида** может повышаться на 5 мг/кг с интервалом от 4 до 7 дней в зависимости от клинического ответа. Для поддерживающей терапии, как правило, достаточно суточной дозы 20 мг/кг у детей и 15 мг/кг у взрослых. Максимальная суточная доза 40 мг/кг массы тела у детей и 30 мг/кг массы тела у взрослых. Суточную дозу принимают в 2 - 3 приема. В связи с длительным периодом полувыведения этосуксимида** при хорошей переносимости всю суточную дозу можно принимать однократно.
Алгоритм постановки диагноза депрессивного эпизода по МКБ-10
В МКБ-10 диагноз депрессивного эпизода устанавливается на основании наличия как минимум двух основных и дополнительных симптомов. Симптомы должны присутствовать ежедневно большую часть времени на протяжении, как минимум, двух недель подряд. Депрессивные эпизоды разделяют по степени тяжести в зависимости от количества основных и дополнительных симптомов, а также нарушения функционирования. При эпизодах легкой и средней степени необходимо уточнять наличие или отсутствие соматических симптомов, а тяжелой степени - психотических. При наличии двух и более депрессивных эпизодов, устанавливается диагноз рекуррентного депрессивного расстройства [546].
Основные симптомы | Дополнительные симптомы | Соматические симптомы | Психотические симптомы |
|---|---|---|---|
сниженное настроение
отчетливое снижение интересов или удовольствия от деятельности, обычно связанной с положительными эмоциями
снижение энергии и повышенная утомляемость | сниженная способность к сосредоточению и вниманию
снижение самооценки и чувство неуверенности в себе
идеи виновности и уничижения (даже при легких депрессиях)
мрачное и пессимистическое видение будущего
идеи или действия, касающиеся самоповреждения или самоубийства
нарушенный сон
нарушенный аппетит | снижение интересов или удовольствия от деятельности, обычно приятной для больного
отсутствие обычной реакции на события или деятельность
пробуждение утром за два или более часа до обычного времени
депрессия тяжелее по утрам
объективные свидетельства заметной психомоторной заторможенности или ажитации (отмеченные или описанные другими лицами)
заметное снижение аппетита
снижение веса (пять или более процентов от веса тела в прошлом месяце)
заметное снижение либидо | бред
галлюцинации
депрессивный ступор
|
Эпизод лёгкой степени: минимум 2 основных симптома и минимум 2 дополнительных симптома;
Эпизод средней степени: минимум 2 основных симптома и минимум 3 дополнительных симптома; значительные затруднения в трудовой и социальной адаптации;
Эпизод тяжелой степени: 3 основных симптома и минимум 4 дополнительных симптома; наличие напряженности или ажитации, либо особенно выраженной заторможенности.
Приложение Б. Алгоритмы действий врача
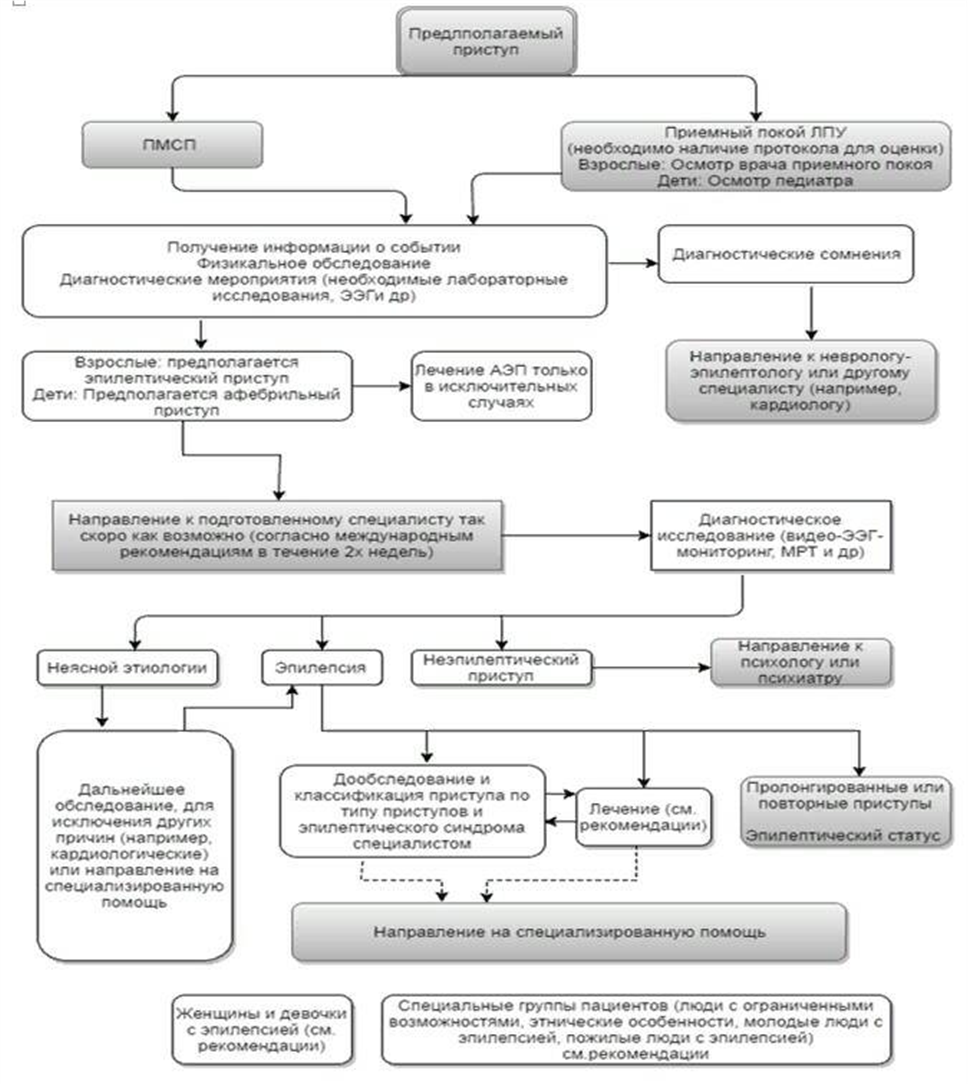
Сокращения: ПМСП – первичная медицинская скорая помощь; ЛПУ – лечебно-профилактическое учреждение; АЭП (ПЭП) – антиэпилептический препарат/противоэпилептический препарат; ЭЭГ – электроэнцефалограмма; МРТ – магнитно-резонансная томография.
Дифференциальный диагноз эпилептических приступов [460]
· Психогенные неэпилептические приступы
· Обморок
· Мигрень
· Гипогликемия
· Панические / тревожные атаки
· Пароксизмальные двигательные расстройства
· Острые дистонические реакции, окулогирный криз
· Гемифациальный спазм
· Парасомнии (REM и non‐REM сна)
· Гипнагогический миоклонус (старты сна)
· Транзиторные ишемические атаки
· Транзиторная глобальная амнезия
· Амнезия
Приложение В. Информация для пациента
Рекомендации по режиму дня
Пациенту с эпилепсией необходимо соблюдать режим сна (ночной сон не менее 8 часов в сутки), избегать нарушений ритма сна, ранних или внезапных пробуждений. Исключить ночной характер труда, так как депривация сна провоцирует возникновение приступов. Необходимо избегать физических и психических перегрузок, правильно чередовать работу и отдых. Более подробные рекомендации даются строго индивидуально с учётом характера приступов пациента.
Нужно ли соблюдать определенную диету?
Питание больных эпилепсией не отличается от питания здоровых людей; оно должно быть полноценным и содержать достаточное количество витаминов и минералов. Нет данных о том, что определенные продукты питания противопоказаны больным, так как могут спровоцировать приступы.
Разработана специальная «кетогенная диета», как метод лечения резистентных и тяжелых форм эпилепсии (например, при синдроме Леннокса - Гасто). Еще в Средние века эпилепсию пытались лечить методом голодания (в Библии содержится упоминание о лечении «молитвой и постом»). Позднее ученые установили, что перестройка обмена веществ при длительном голодании может привести к уменьшению частоты приступов. Однако, лечение эпилепсии голоданием не получило распространения, так как этот метод тяжело переносится и опасен, особенно у детей. В связи с этим ученые пытались найти другие методы диетического воздействия (кроме голодания), которые приводили бы к изменениям обмена веществ в организме, подобным изменениям, возникающим при голодании. Кетогенная диета была изобретена в 20-е гг. XX века в США. Диета не требует длительного голодания, а заключается в ограничении углеводной пищи и белков и преобладания в рационе питания жиров. При переваривании пищи жиры превращаются в специфические продукты обмена — кетоновые тела, которые попадают в головной мозг и обеспечивают противосудорожный эффект. Как и другие методы лечения, кетогенная диета имеет серьезные побочные эффекты и противопоказания, и должна применяться только под контролем врача в специализированных центрах. Для каждого пациента рацион питания рассчитывается индивидуально.
Какие меры безопасности нужно соблюдать?
Люди, страдающие эпилепсией, должны стараться вести обычный активный образ жизни; однако, если, несмотря на лечение, сохраняются приступы с нарушением сознания, необходимо соблюдать простые правила безопасности, уменьшающие вероятность травмы во время приступа.
· Пациент не должен находиться без страховки на высоте, у края платформ железнодорожных станций, около огня, вблизи водоемов, у движущихся механизмов.
· Пациенту с эпилепсией нельзя принимать ванну, поскольку существует риск утопления в воде во время развития приступа.
· Пациенту с эпилепсией необходимо избегать отдельных видов спорта, связанных с повышенной травматизацией (бокс, прочие виды боевых искусств, спортивная гимнастика, конный спорт, погружение с аквалангом, скалолазание, альпинизм, дельтапланеризм, парашютный спорт).
· При катании на велосипеде необходимо надевать шлем и другие защитные средства (наколенники и налокотники). Не следует выезжать на проезжую часть. Наиболее благоприятное место для велосипедной прогулки ― городской парк.
· Пациент с эпилепсией не должен управлять автомобилем, мотоциклом и др. транспортными средствами.
· При фотосенситивных формах эпилепсии необходимо использовать поляризующие очки.
· Избегать повышенной инсоляции.
· Исключить употребление алкоголя, энергетических напитков, ограничить употребление кофе.
Кому пациент должен рассказать о своем заболевании?
Пациентам обычно советуют рассказать о своем заболевании коллегам по работе, учителям в школе, особенно при плохо контролируемых приступах, для того, чтобы в случае возникновения приступа могла быть оказана своевременная помощь. Целесообразно ношение специальной карточки, браслета или медальона с информацией о заболевании. Информация о заболевании часто требуется при приеме на работу и ее не рекомендуется скрывать, даже при возможных препятствиях в получении работы (особенно, если существует вероятность развития приступа на работе, если необходимы особые условия работы, связанные с болезнью, если существует опасность для здоровья больного и окружающих людей в случае развития приступа).
Компьютер и телевизор
У некоторых людей с эпилепсией (приблизительно в 5–15% случаев) приступы могут провоцироваться ритмичным мельканием света. Это явление названо фотосенситивностью (фоточувствительностью) и выявляется во время исследования ЭЭГ, когда пациент смотрит на лампочку, мелькающую с разной частотой. Фотосенситивность в 2,5 раза чаще встречается у женщин, чем у мужчин. У пациентов с фотосенситивностью приступы могут провоцироваться просмотром телевизора, компьютерными играми, цветомузыкой на дискотеке. Именно пациентам с фотосенситивностью следует ограничить просмотр телевизора и работу на компьютере.
Однако источником световых мельканий могут служить не только телевизоры и компьютеры, но и природные явления (яркие блики на воде, искрящийся снег в солнечный день, чередование света и тени и др.)
Даже детей с фоточувствительностью не рекомендуется совсем лишать просмотра телевизора, так как это снижает качество жизни ребенка, становится причиной переживаний.
Правила, которые необходимо соблюдать ребенку с эпилепсией при просмотре телепередач:
· Ребенок не должен смотреть телевизор более 1 – 1,5 часа.
· Расстояние от ребенка до телевизора должно быть максимальным, насколько позволяет комната (но не менее 2 метров).
· Обязательно дополнительное освещение комнаты для уменьшения светового контраста.
· Предпочтение отдается телевизорам с меньшим размером экрана.
· Телевизор должен быть цветным с нерезко отрегулированным контрастом и высокой частотой разверстки (100 Гц).
· Для управления телевизором нужно пользоваться дистанционным пультом.
· Чтобы уменьшить эффект мерцания при просмотре мелькающих картинок, вспышек, калейдоскопических съемок нужно закрыть один глаз.
· Не следует смотреть телевизор, если ребенок не выспался, утомлен или чувствует себя не достаточно хорошо.
Работа на компьютере — также возможный провоцирующий фактор приступов у людей с фотосенситивной эпилепсией, однако, полный отказ от компьютера в настоящее время вызовет целый ряд неблагоприятных психологических и социальных последствий в связи с тем, что:
· Навыки работы на компьютере нужны при устройстве на работу в различных сферах.
· В тех случаях, когда предпочтительна работа в домашних условиях (например, у больных с сопутствующими двигательными нарушениями), компьютер — это возможность профессиональной реализации для людей многих профессий (творческие профессии, переводчики, специалисты в области информационных технологий и др.). Возможность работы на компьютере может определить выбор профессии у многих больных, имеющих другие ограничения.
· Для детей существует множество развивающих и обучающих компьютерных программ, которые позволят более эффективно усвоить учебный материал; использование компьютерных программ может быть особенно полезно детям, находящимся на домашнем обучении.
· Компьютер значительно расширяет возможности общения как у детей, так и у взрослых с эпилепсией.
Таким образом, лишение пациента с эпилепсией компьютера может означать не только ограничения возможностей для отдыха и развлечений, но также ограничение социальной активности, возможностей для творческой и профессиональной реализации и обучения. Для того чтобы не лишать больного возможности работы на компьютере, нужно соблюдать ряд правил.
Правила, которые необходимо соблюдать пациенту с эпилепсией во время работы или игры на компьютере:
· Продолжительность работы/игры на компьютере не должна превышать 1–1,5 часа с обязательным перерывом через каждые 30 минут на 10–15 минут, которые необходимы для отдыха глаз.
· Расстояние от пациента до монитора составляет 70 см (вытянутая рука взрослого с вытянутыми пальцами); расстояние от глаз до монитора должно быть не менее 35 см для 14-дюймовых экранов.
· Обязательно дополнительное освещение комнаты для уменьшения светового контраста.
· На монитор не должны попадать блики от окон и других источников света.
· Монитор предпочтительно выбирать жидкокристаллический, или с большой разрешающей способностью; отдавать предпочтение стандарту SVGA с частотой разверстки не менее 60 Гц.
· Экран монитора должен быть чистым, параметры изображения следует правильно отрегулировать.
· Нельзя рассматривать мелкие детали изображения с близкого расстояния.
· Необходимо убрать из поля зрения другие мониторы и телевизоры.
· Не следует работать/играть на компьютере, если пациент не выспался, утомлен или плохо себя чувствует.
Перечисленные рекомендации подходят как для детей, так и для взрослых с эпилепсией, и будут также полезны здоровым людям.
Опасно ли для пациентов с эпилепсией посещение дискотек?
Этот вопрос, вероятно, наиболее актуален для подростков и пациентов молодого возраста. Известно, что светомузыкальные эффекты могут спровоцировать приступ у фоточувствительных людей; однако, опасны не все элементы светомузыки, а только стробоскопические (часто мерцающие) яркие блики в темном помещении. Яркость и частота мелькания — важные факторы, влияющие на вероятность развития приступа.
Важно отметить, что с посещением дискотек связаны и другие факторы, провоцирующие развитие приступов — недосыпание, усталость и прием алкоголя.
Выбор профессии
Ограничения в выборе профессии у пациентов с эпилепсией связаны с существующим риском возникновения судорожных приступов в ситуациях, когда они могут причинить вред пациенту или подвергнуть опасности жизнь других людей. Людям с эпилептическими приступами нельзя управлять транспортом (правила, связанные с вождением автомобиля, зависят от законодательных норм страны; более жёсткие ограничения установлены в отношении пассажирского транспорта), работать у незащищенных механизмов, на высоте, вблизи водоемов, служить в армии и на военно-морском флоте, в полиции, пожарных частях, в тюрьмах, охране, на скорой помощи. Также для пациента с эпилепсией представляет потенциальную опасность работа с движущимися механизмами, с ценными хрупкими объектами, с химикатами.
В целом, на способность человека к выполнению какой-либо деятельности влияют тип эпилепсии, тяжесть заболевания, наличие сопутствующих физических или интеллектуальных нарушений и степень контроля приступов.
Работа в сменном режиме обычно не вредна для пациента, если существует возможность полноценного сна и регулярного приема лекарств в соответствии с назначениями врача.
Важно помнить, что диагноз «эпилепсия» не должен служить препятствием для получения образования и успешной реализации в выбранной профессиональной сфере. Как и любой другой человек, пациент с эпилепсией сможет выбрать сферу деятельности, в которой он сможет реализовать свои способности наилучшим образом и стать полноценным членом общества.
Приложение Г1-ГN. Шкалы оценки, вопросники и другие оценочные инструменты состояния пациента, приведенные в клинических рекомендациях
Приложение Г1. Неврологический опросник депрессивного расстройства при эпилепсии
Название на русском языке: «Неврологический опросник депрессивного расстройства при эпилепсии»
Оригинальное название (если есть): Neurological Disorders Depression Inventory for Epilepsy – NDDI-E
Источник (официальный сайт разработчиков, публикация с валидацией):
Оригинальная версия: Gilliam F. G. et al. Rapid detection of major depression in epilepsy: a multicentre study //The Lancet Neurology. – 2006. – Т. 5. – №. 5. – С. 399-405.
Русскоязычная версия: Zinchuk M. et al. Validation of the Russian version of neurological disorders depression inventory for epilepsy (NDDI-E) // Epilepsy Behav. Academic Press Inc., 2020. Vol. 113, № 107549.
Тип (подчеркнуть):
- шкала оценки
- индекс
- вопросник
- другое (уточнить):
Назначение: Опросник предназначен для скрининга текущего депрессивного эпизода у пациентов с эпилепсией 18 лет и старше.
Содержание (шаблон):
Для каждого из следующих утверждений обведите число, которое лучше всего описывает Вас на протяжении последних двух недель, включая сегодня.
| Все время или часто | Иногда | Редко | Никогда |
|---|---|---|---|---|
Всё дается с трудом | 4 | 3 | 2 | 1 |
Я все делаю не так | 4 | 3 | 2 | 1 |
Я чувствую вину | 4 | 3 | 2 | 1 |
Мне лучше было бы умереть | 4 | 3 | 2 | 1 |
Я чувствую разочарование и опустошённость | 4 | 3 | 2 | 1 |
Мне трудно получить удовольствие | 4 | 3 | 2 | 1 |
Ключ (интерпретация): Оптимальная точка для выявления текущего депрессивного эпизода – больше 12 баллов (Se – 88,16%, Sp – 81,82%).
Приложение Г2. Шкала Генерализованного тревожного расстройства (ГТР-7)
Название на русском языке: Шкала Генерализованного тревожного расстройства (ГТР-7)
Оригинальное название: General Anxiety Disorder-7
Источник: Spitzer RL, Kroenke K, Williams JB, Löwe B. A brief measure for assessing generalized anxiety disorder: the GAD-7. Arch Intern Med. 2006;166:1092-7.
Micoulaud-Franchi J.A. et al. Rapid detection of generalized anxiety disorder and major depression in epilepsy: Validation of the GAD-7 as a complementary tool to the NDDI-E in a French sample // Epilepsy Behav. Academic Press Inc., 2016. Vol. 57, № Pt A. P. 211–216.
Тип (подчеркнуть):
- шкала оценки
- индекс
- вопросник
- другое (уточнить):
Назначение: Опросник предназначен для скрининга тревожных расстройств у пациентов с эпилепсией 18 лет и старше
Содержание (шаблон):
Опросник ГТР - 7 | ||||
|---|---|---|---|---|
Как часто за последние 2 недели Вас беспокоили следующие проблемы? | Никогда | Несколько дней | Более половины дней | Почти каждый день |
1. Повышенная нервная возбудимость, беспокойство или раздражительность | 0 | 1 | 2 | 3 |
2.Неспособность справиться с волнением | 0 | 1 | 2 | 3 |
3.Чрезмерное беспокойство по разному поводу | 0 | 1 | 2 | 3 |
4.Неспособность расслабляться | 0 | 1 | 2 | 3 |
5.Крайняя степень беспокойства: «не могу найти себе места» | 0 | 1 | 2 | 3 |
6.Легко поддаюсь чувству беспокойства или раздражительности | 0 | 1 | 2 | 3 |
7. Опасение чего-то страшного | 0 | 1 | 2 | 3 |
Ключ: Оптимальная точка для выявления любого тревожного расстройства - больше 8 баллов.
Приложение Г3. Госпитальная шкала тревоги и депрессии (HADS)
Название на русском языке: Госпитальная шкала тревоги и депрессии (HADS)
Оригинальное название: Hospital Anxiety and Depression Scale (HADS)
Источник: Wiglusz MS, Landowski J, Michalak L, Cubała WJ. Validation of the Hospital Anxiety and Depression Scale in patients with epilepsy. Epilepsy Behav. 2016 May;58:97-101.
Тип (подчеркнуть):
- шкала оценки
- индекс
- вопросник
- другое (уточнить):
Назначение: Опросник предназначен для скрининга тревожных расстройств и депрессивного расстройства у пациентов с эпилепсией 18 лет и старше.
Содержание (шаблон):
Прочитайте внимательно каждое утверждение и отметьте ответ, который в наибольшей степени соответствует тому, как Вы себя чувствовали на прошлой неделе. Не раздумывайте слишком долго над каждым утверждением. Ваша первая реакция всегда будет более верной.
Д | Т |
| Д | Т |
|
|---|---|---|---|---|---|
|
| Я испытываю напряжение, мне не по себе |
|
| Мне кажется, что я стал все делать очень медленно |
| 3 | все время | 3 |
| практически все время |
| 2 | часто | 2 |
| часто |
| 1 | время от времени, иногда | 1 |
| иногда |
| 0 | совсем не испытываю | 0 |
| совсем нет |
|
| То, что приносило мне большое удовольствие, и сейчас вызывает у меня такое же чувство |
|
| Я испытываю внутреннее напряжение или дрожь |
0 |
| определенно, это так |
| 0 | совсем не испытываю |
1 |
| наверное, это так |
| 1 | иногда |
2 |
| лишь в очень малой степени это так |
| 2 | часто |
3 |
| это совсем не так |
| 3 | очень часто |
|
| Мне страшно. Кажется, будто что-то ужасное может вот-вот случиться |
|
|
Я не слежу за своей внешностью |
| 3 | определенно это так, и страх очень сильный | 3 |
| определенно это так |
| 2 | да, это так, но страх не очень сильный | 2 |
| я не уделяю этому столько времени, сколько нужно |
| 1 | иногда, но это меня не беспокоит | 1 |
| может быть, я стал меньше уделять этому внимания |
| 0 | Совсем не испытываю | 0 |
| я слежу за собой так же, как и раньше |
|
| Я способен рассмеяться и увидеть в том или ином событии смешное |
|
| Я испытываю неусидчивость, словно мне постоянно нужно двигаться |
0 |
| определенно, это так |
| 3 | определенно, это так |
1 |
| наверное, это так |
| 2 | наверное, это так |
2 |
| лишь в очень малой степени это так |
| 1 | лишь в очень малой степени это так |
3 |
| совсем не способен |
| 0 | совсем не испытываю |
|
| Беспокойные мысли крутятся у меня в голове |
|
| Я считаю, что мои дела (занятия, увлечения) могут принести мне чувство удовлетворения |
| 3 | постоянно | 0 |
| точно так, как и обычно |
| 2 | большую часть времени | 1 |
| да, но не в той степени, как раньше |
| 1 | время от времени | 2 |
| значительно меньше, чем раньше |
| 0 | только иногда | 3 |
| совсем так не считаю |
|
| Я чувствую себя бодрым |
|
| У меня бывает внезапное чувство паники |
3 |
| совсем не чувствую |
| 3 | действительно, очень часто |
2 |
| очень редко |
| 2 | довольно часто |
1 |
| иногда |
| 1 | не так уж часто |
0 |
| практически все время |
| 0 | совсем не бывает |
|
| Я легко могу сесть и расслабиться |
|
| Я могу получить удовольствие от хорошей книги, фильма, радио- или телепрограммы |
| 0 | определенно, это так | 0 |
| часто |
| 1 | наверное, это так | 1 |
| иногда |
| 2 | лишь изредка это так | 2 |
| редко |
| 3 | совсем не могу | 3 |
| очень редко |
Ключ: Для интерпретации необходимо суммировать баллы по каждой подшкале (Д и Т) в отдельности:
0 - 7 баллов норма: (отсутствие достоверно выраженных симптомов тревоги и депрессии)
8 - 10 баллов: субклинически выраженная тревога /депрессия
11 баллов и выше: клинически выраженная тревога / депрессия
Приложение Г4. «Неврологический опросник депрессивного расстройства при эпилепсии» (Neurological Disorders Depression Inventory for Epilepsy – NDDI-E)
ФИО: _________________________________________________________
Дата: _____________________________________________________________
Для каждого из следующих утверждений обведите число, которое лучше всего описывает Вас на протяжении последних двух недель, включая сегодня.
| Все время или часто | Иногда | Редко | Никогда |
|---|---|---|---|---|
Всё дается с трудом | 4 | 3 | 2 | 1 |
Я все делаю не так | 4 | 3 | 2 | 1 |
Я чувствую вину | 4 | 3 | 2 | 1 |
Мне лучше было бы умереть | 4 | 3 | 2 | 1 |
Я чувствую разочарование и опустошённость | 4 | 3 | 2 | 1 |
Мне трудно получить удовольствие | 4 | 3 | 2 | 1 |
Ключ: Оптимальная точка для текущего депрессивного эпизода - больше 12 баллов.