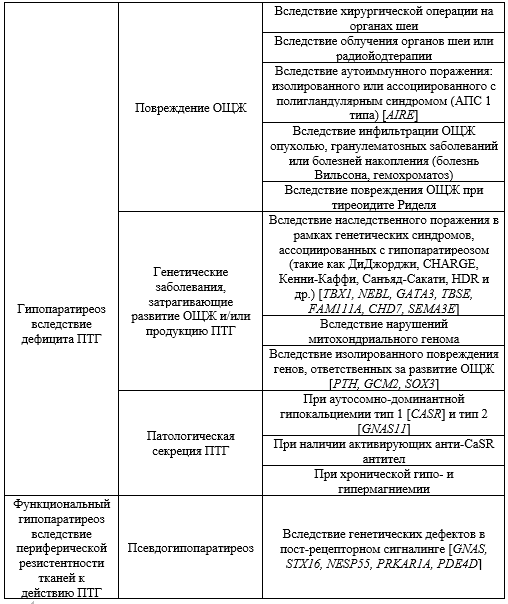
7.1 Лечение острой гипокальциемии
Для купирования острой гипокальциемии рекомендуется установка центрального венозного катетера, что позволит предотвратить склерозирование периферических вен вследствие инфузии препаратов кальция. Предпочтительно использование кальция глюконата**, так как кальция хлорид имеет серьезные осложнения в виде некроза мягких тканей, в случае выхода раствора из сосудистого русла.
Для купирования острой гипокальциемии используется внутривенное введение препаратов кальция в 2 этапа: 1-2 ампулы 10% раствора кальция глюконата**, содержащие 90-180 мг элементарного кальция соответственно, в 50 мл 0,9% раствора натрия хлорида** (или в 5% раствора декстрозы**) в течение 10-20 минут; последующее медленное внутривенное введение кальция глюконата** 100 мл (10 ампул) со скоростью 0,5-1,5 мг/кг/час в 1000 мл 0,9% раствора натрия хлорида** (или 5% раствора декстрозы) в течение, в среднем, 8-10 часов.
В зависимости от ситуации могут быть использованы следующие варианты парентерального введения препаратов кальция:
Введение кальция глюконата** внутривенно болюсно в количестве 20-60 мл – быстро без разведения 0,9% раствором натрия хлорида** (или в 5% раствора декстрозы). Метод используется при выраженных симптомах гипокальцемии (пациенты с клинической картиной «страха смерти» или в бессознательном состоянии). Доза вводимого кальция определяется по появлению диспепсических жалоб.
Введение половины дозы кальция глюконата** внутривенно болюсно без разведения (40-50 мл 10% раствора кальция глюконата**), остальная доза кальция (50-60 мл 10% раствора кальция глюконата**) вводится внутривенно медленно в разведенном состоянии (0,9% раствора натрия хлорида** или 5% декстрозы**) со скоростью для инфузомата 0,5-1,5 мкг/кг/час. Данный способ введения является самым частым для купирования острой гипокальцемии, позволяющим быстро нормализовать клиническое состояние пациента.
Введение всей дозы парентеральных препаратов кальция (80-100 мл 10% раствора кальция глюконата**) разведенного в растворе (0,9% натрия хлорида** или 5% декстрозы**). Метод используется преимущественно для поддержания адекватного уровня кальция с целью профилактики развития острой гипокальцемии.
Парентеральное введение препаратов кальция всегда прекращается при появлении диспептических жалоб (тошнота, рвота).
Внутривенное введение кальция требует осторожности у больных с гипокалиемией и у пациентов, принимающих дигоксин**, в связи с повышенным риском аритмий.
Одновременно назначаются пероральные препараты кальция и препараты витамина D и его аналогов (альфакальцидол**, кальцитриол**). Цель терапии – купирование симптомов острой гипокальциемии вследствие гипопаратиреоза и нормализация показателей общего и ионизированного кальция на нижней границе референсных значений или несколько ниже в отсутствие клинических симптомов гипокальциемии. Для коррекции терапии необходим частый контроль уровня кальция крови (каждые 6-12 часов в начале лечения, после стабилизации состояния пациента – каждые 24 часа).
Стоить отметить, что при наличии у пациента выраженной гипомагниемии показана ее коррекция с использованием как пероральных препаратов (препараты комбинации различных препаратов магния 300-400 мг/сут), так и внутривенных форм – внутривенно струйно 2 грамма магния сульфата** в течение 10-20 минут, внутривенно капельно 25% раствор магния сульфата** 2-4 грамма в 150-200 мл физиологического раствора натрия хлорида**.
Алгоритм купирования острой гипокальциемии вследствие гипопаратиреоза:
1) Кальция глюконат** 20-60 мл в/в болюсно, и/или 1-2 ампулы 10% раствора кальция глюконата** (90-180 мг элементарного кальция соответственно) в 50-100 мл 0,9% раствора натрия хлорида** (или 5% водного раствора декстрозы**) в/в в течение 10-20 минут с одновременным назначением пероральных препаратов кальция и препаратов витамина D и его аналогов (альфакальцидол**, кальцитриол**). При необходимости может быть продолжена инфузионная терапия кальция глюконатом** по схеме: 10 ампул 10% раствора кальция глюконата** (900 мг элементарного кальция) в 1000 мл 0,9% раствора NaCl** (или 5% раствора декстрозы**) со скоростью 50 мл/час (в среднем в течение 8-10 часов).
2) Препараты витамина D и его аналогов (альфакальцидол** в среднем 3-3,5 мкг/сут и/или кальцитриол** 1,5-2 мкг/сут перорально) в сочетании с препаратами кальция в среднем 3000 мг по элементарному кальцию внутрь на 3-6 приемов в сутки во время или сразу после приема пищи). При сохранении гипокальциемии могут быть применены более высокие дозы под контролем кальция и фосфора крови.
7.2 Ведение беременности при гипопаратиреозе
Физиологические аспекты регуляции минерального обмена во время беременности
К одному из основных компенсаторных механизмов, обеспечивающих плод достаточным количеством кальция во время беременности, относится усиление его абсорбции в кишечнике матери. В период лактации адекватная концентрация кальция в грудном молоке достигается, в основном, за счет усиления резорбтивных процессов в костной ткани. Эти изменения связаны с увеличением продукции таких ключевых гормонов как кальцитриол и ПТГ-подобный пептид (ПТГпП).
Увеличение синтеза кальцитриола наблюдается с I триместра и к концу беременности его уровень составляет до 2-3 норм. Регуляция метаболизма витамина D у беременной женщины в основном зависит от активности 1α-гидроксилазы почек, находящейся под контролем эстрогенов, пролактина и человеческого плацентарного лактогена. При этом уровень 25(ОН)D остается стабильным, несмотря на усиление его конверсии в активную форму. Во многих тканях организма, включая молочные железы, децидуальную оболочку, плаценту и др., увеличивается синтез ПТГпП. Уровень ПТГпП возрастает с 3 по 13 неделю гестации, и более чем втрое превышает показатель до беременности. Именно в эти периоды ПТГпП действует как эндокринный фактор, регулирующий минеральный и костный гомеостаз независимо от ПТГ. ПТГпП так же, как и ПТГ, способен стимулировать резорбцию костной ткани, реабсорбцию кальция в почках, что в комплексе обеспечивает насыщение грудного молока необходимым для новорожденного количеством кальция.
Продукция ПТГ ОЩЖ при наступлении беременности снижается и его уровень в крови держится в низко-нормальном диапазоне или даже ниже популяционных референсных показателей в I триместре, что связано с гестационным повышением продукции ПТГпП и кальцитриола с 3-ей недели беременности. Таким образом, ПТГ, как регулятор фосфорно-кальциевого обмена, теряет свою доминирующую роль в этот период и только к концу III-го триместра его продукция восстанавливается.
Кальцитонин – гормон-антагонист ПТГ, продуцируемый С-клетками щитовидной железы, а в периоды беременности и лактации – молочными железами и плацентой. Повышение уровня кальцитонина во время беременности и лактации отмечено также у пациенток, ранее перенесших тотальную тиреоидэктомию. Увеличение синтеза этого гормона во время беременности реализуется за счет эффектов эстрогенов (эстрадиола, эстрона и эстриола) и направлено на подавление активной деминерализации скелета матери.
Во время беременности и лактации за счет увеличения объема циркулирующей крови и гемодилюции снижается уровень общего кальция сыворотки крови, в то время как уровни альбумин-скорректированного и ионизированного кальция остаются неизменными. Уровень кальция крови плода, как правило, оказывается несколько выше показателей кальциемии у матери.
Исследования по ведению беременности на фоне хронического гипопаратиреоза ограничены небольшими выборками (чаще описание серии клинических случаев). Тем не менее, экспертное сообщество выделяет ключевые положения по тактике ведения и лечения данного заболевания при гестации.
Основные принципы по ведению беременности у пациентки с хроническим гипопаратиреозом:
- оптимальным считается поддержание уровней ионизированного и альбумин-скорректированного кальция крови в пределах нижне-нормального диапазона (для альбумин-скорректированного кальция в пределах 2,1-2,3 ммоль/л, для ионизированного кальция – в пределах 1,05-1,15 ммоль/л) во избежание неблагоприятного воздействия на развитие ОЩЖ плода. Для расчета уровня альбумин-скорректированного кальция применяют формулу: альбумин-скорректированный кальций плазмы (с поправкой) (ммоль/л) = измеренный уровень общего кальция плазмы (ммоль/л) + 0,02 × (40 - измеренный уровень альбумина плазмы (г/л));
- в период гестации следует проводить мониторинг показателей фосфорно-кальциевого обмена (исследование уровня общего кальция в крови, исследование уровня альбумина в крови, исследование уровня ионизированного кальция в крови, исследование уровня фосфора в крови) с частотой не реже чем 1 раз в 4-6 недель для профилактики гипо- и гиперкальциемии;
- в случае изменения терапии может потребоваться более частый контроль, в среднем 1 раз в 7-14 дней до достижения целевых показателей кальциемии;
- следует поддерживать уровни фосфора, магния и 25(ОН)D в пределах референсного диапазона;
- следует прекратить лечение тиазидами/тиазидоподобными диуретиками на период беременности и лактации;
- следует прекратить лечение препаратами паратиреоидных гормонов и их аналогами на период беременности и лактации;
- пациентки с хроническим гипопаратиреозом нуждаются в обучении, направленном на повышение информированности о симптоматике гипо- и гиперкальциемии.
В представленных исследованиях по лечению хронического гипопаратиреоза во время беременности у женщин наблюдались значительные изменения потребности в препаратах кальция и препаратах витамина D и его аналогов (чаще кальцитриола**, реже #альфакальцидола) в зависимости от срока гестации и периода лактации. Средние дозы препаратов кальция достигали 2000-3000 мг в сутки, дозы кальцитриола** – 1-2 мкг в сутки, дозы #альфакальцидола – в среднем 2 мкг в сутки. Лабораторный контроль проводился каждые 4-6 недель в течение всей беременности для обеспечения адекватного управления состоянием и корректировки дозировок при необходимости. Важно отметить, что несмотря на то, что беременность и период лактации заявлены в противопоказаниях для кальцитриола**/#альфакальцидола**, альтернатив для лечения хронического гипопаратиреоза в период гестации не существуют. В доступных исследованиях продемонстрирована безопасность применения данных лекарственных препаратов в указанных дозах [180-182].
7.3. Аутоиммунный гипопаратиреоз
Аутоиммунный гипопаратиреоз – вторая по распространенности форма гипопаратиреоза, обусловленная иммуно-опосредованным разрушением клеток ОЩЖ. Он может быть изолированным заболеванием, однако значительно чаще встречается в рамках наследственного АПС 1-го типа, также известного как кандидо-эктодермальная дистрофия (Autoimmune Polyendocrinopathy with Candidiasis and Ectodermal Dystrophy – APECED) [14]. Распространенность данного заболевания оценена в 1:100 000 населения в большинстве стран, большая распространенность отмечена в таких странах как Финляндия (1:25000) и Сардиния (1:14000) и среди персидских евреев в Израиле (1:9000) [166].
АПС 1-го типа – моногенное аутосомно-рецессивное заболевание, в основе которого лежит нарушение структуры гена аутоиммунного регулятора (AIRE). Ген AIRE располагается на длинном плече 21-й хромосомы, кодирует ядерный фактор транскрипции и играет одну из ключевых ролей в формировании иммунотолерантности. В основе патогенеза лежит аутоиммунная деструкция различных эндокринных желез [167]. Для АПС 1-го типа характерна классическая триада: слизисто-кожный кандидоз, гипопаратиреоз, хроническая надпочечниковая недостаточность. Заболевание дебютирует, как правило, в детском возрасте. Более чем у 80% пациентов с АПС 1 типа гипопаратиреоз является единственным эндокринным проявлением заболевания [167].
В подавляющем большинстве случаев первым проявлением становится слизисто-кожный кандидоз, развивающийся в первые 10 лет жизни, чаще в возрасте около 2 лет. На фоне слизисто-кожного кандидоза у 84% пациентов появляется гипопаратиреоз, при этом в 88% случаев он дебютирует в возрасте до 10 лет. К другим компонентам синдрома относятся сахарный диабет 1-го типа, тиреоидит Хашимото, целиакия, витилиго, аутоиммунный гепатит, гипогонадизм и другие [16].
Компоненты АПС 1 типа:
- Главные компоненты:
- слизисто-кожный кандидоз;
- гипопаратиреоз;
- первичная надпочечниковая недостаточность.
- Другие возможные проявления:
- кератит;
- аутоиммунный гепатит;
- первичная овариальная дисфункция;
- гипоплазия зубной эмали;
- энтеропатия с хронической диареей или запорами;
- фотофобия;
- периодический жар с сыпью;
- пневмонит;
- нефрит;
- панкреатит;
- сахарный диабет 1 типа;
- функциональная аспления;
- целиакия;
- тиреоидит;
- ретинит;
- аплазия красного ростка костного мозга;
- полиартрит.
В настоящее время остается неизвестным, что выступает мишенью для аутоиммунного повреждения клеток ОЩЖ. В литературе рассматривается несколько антигенов, аутоантитела к которым могут быть ответственны за развитие гипопаратиреоза. В 1996 году Y. Li и соавт. впервые выявили активирующие антитела к CаSR. В исследование были включены 25 пациентов, среди которых у 17 причиной гипопаратиреоза был АПС 1-го типа, а у 8 человек отмечалось сочетание гипопаратиреоза и аутоиммунного тиреоидита. В результате у 56% пациентов с гипопаратиреозом определялись высокие титры антител к CаSR, тогда как в группе контроля эти антитела не были найдены ни у одного человека [168]. Однако в ряде других работ корреляции между активирующими антителами к CаSR и гипопаратиреозом у пациентов с АПС 1-го типа выявлено не было [169]. Антитела к CаSR, но обладающие блокирующей способностью, также были обнаружены при аутоиммунной гипокальциурической гиперкальциемии [170].
NALP5 – мультипротеиновый комплекс, активирующий внутриклеточные киназы и синтез провоспалительных цитокинов, включает в себя: NACНT (neuronal apoptosis inhibitor protein), C2TA (MHC class 2 transcription activator), HET-E (incompatibility locus protein from Podospora anserina) и TP1 (telomerase-associated protein), впервые обнаружен M. Alimohammadi и соавт. в качестве специфического антигена ОЩЖ. Всего было обследовано 87 пациентов с АПС 1-го типа, у 73 из которых отмечался гипопаратиреоз. Среди пациентов с гипопаратиреозом антитела к NALP5 идентифицированы в 49% наблюдений, у пациентов с АПС 1-го типа без снижения функции ОЩЖ данные антитела не были обнаружены ни у одного пациента [171]. Подобные результаты получены в работе A. Meloni и соавт., в которой антитела к NALP5 выявлены у 64,3% пациентов с гипопаратиреозом и не были обнаружены у пациентов без гипопаратиреоза [172]. Различные формы аутоиммунного гипопаратиреоза представлены в таблице 8.
Таблица 8. Аутоиммунные формы гипопаратиреоза.
Возраст манифестации | Ассоциированные заболевания | Генетика | Антитела | Заболевания/ синдромы |
5-20 лет | Слизисто-кожный кандидоз и/или болезнь Аддисона | Мутации в AIRE | NALP5Abs, IFNɷAbs, TPHAbs, AADC, THAbs, ACA, 21-OHAbs | АПС 1 типа |
Взрослые | Аутоиммунные заболевания ЩЖ | HLA (?) | CaSRAbs (?) | АПС 3 типа |
Взрослые | Любые другие аутоиммунные заболевания (исключая слизисто-кожный кандидоз, болезнь Аддисона и аутоиммунные заболевания ЩЖ) | HLA (?) | CaSRAbs (?) | АПС 4 типа |
Взрослые | Нет | HLA (?) | CaSRAbs (?) | Изолирован. аутоиммунн. ГипоПТ |
Характеристика основных синдромов в рамках которых встречается аутоиммунный гипопаратиреоз представлена в таблице 9.
Таблица 9. Классификация и характеристика АПС.
Характеристика | АПС 1 | АПС 2 | IPEX |
Основные проявления | Болезнь Аддисона, гипопаратиреоз, хронический слизисто-кожный кандидоз | Болезнь Аддисона, аутоиммунные заболевания ЩЖ, сахарный диабет 1 типа | Аутоиммунная энтеропатия, неонатальный сахарный диабет 1 типа, экзема |
Другие, ассоциированные проявления | Первичный гипогонадизм, аутоиммунные заболевания ЩЖ, сахарный диабет 1 типа, гастрит, энтерит с мальабсорбцией, гепатит, панкреатит, пневмонит, нефрит, витилиго, алопеция, дистрофия ногтей, гипоплазия зубной эмали, кератит, ретинит | Аутоиммунный гастрит, алопеция, витилиго, целиакия, первичный гипогонадизм | Аутоиммунные заболевания ЩЖ, гемолитическая анемия, тромбоцитопения |
Типичный возраст манифестации | Детство, подростковый период | Подростковый период, взрослый | Младенчество |
Распространенность | 1:100 000 | 1:1000 | 1:1 000 000 |
Лечение | Заместительная гормональная, противогрибковая, иммунносупрессивная терапия при гепатите, мальабсорбции, нефрите, пневмоните и кератите | Заместительная гормональная терапия | Заместительная гормональная терапия, трансплантация костного мозга |
Осложнения, включая смерть | Адреналовый и гипокальциемический кризы, рак ротовой полости и пищевода | Адреналовый криз, осложнения сахарного диабета | Инфекции |
Гены и тип наследования | AIRE, аутосомно-рецессивно или доминантно | Полигенетический: MHC и другие | FOXP3, Х-связанный |
Иммунный фенотип | Аутоантитела к интерферону-ω и интерферону-α (>95%), орган-специфические внутриклеточные белки | Аутоантитела к 21-гидроксилазе, GAD65, IA-2, тиротропиновому рецептору, TPO | Аутоантитела к GAD65, лимфоцитоз, эозинофилия, гиперпродукция цитокинов, гипер IgE |
IPEX – Х-связанный иммунодисрегуляторная полиэндокринопатия и энтеропатия, MHC – главный комплекс гистосовместимости, GAD65 – glutamic acid decarboxylase 65; IA-2 – islet antigen 2; TPO – тиреоидная пероксидаза.
Интерфероны (ИФН) впервые были открыты в 1957 г. как агенты, защищающие клетки от вирусной инфекции. Выделяют две группы ИФН: к группе ИФН 1-го типа относятся: ИФН-α, ИФН-β, ИФН-ε, ИФН-κ, ИФН-ω, ИФН-δ, ИФН-τ, а к ИФН 2-го типа только ИФН-γ [173]. При проведении исследования пациентов с генетически подтвержденным диагнозом АПС 1 типа, а также АПС 2 типа и изолированным аутоиммунным поражением органов-мишеней было получено, что у пациентов с АПС 1 типа были выявлены высокие титры антител к большинству подтипов ИФН-α, особенно к ИФН-α2 и ИФН-ω (у 100% пациентов). Ни у одного из пациентов с АПС 2-го типа или изолированными аутоиммунными заболеваниями эти антитела обнаружены не были. Таким образом, антитела к ИНФ-ω оказались высокоспецифичными для синдрома АПС 1-го типа независимо от спектра манифестировавших компонентов [110].
Присутствие антител к ИФН-ω является диагностическим критерием АПС 1-го типа. Секвенирование гена AIRE по-прежнему остается важным этапом диагностики этого синдрома, поскольку позволяет проводить генетическое консультирование и осуществлять планирование семьи. Алгоритм диагностики различных форм аутоиммунного гипопаратиреоза представлен в блок-схеме 1.
Блок-схема 1. Алгоритм диагностики различных форм аутоиммунного гипопаратиреоза.

7.4. Гипопаратиреоз в рамках наследственных синдромов
Синдром ДиДжорджи
Синдром ДиДжорджи является следствием неправильного развития органов-производных третьего и четвертого жаберных карманов. Встречается он у 1 из 4000-5000 новорожденных, и в большинстве случаев (70-80%) является следствием гетерозиготной микроделеции в 22q11.21-q11.23 участках. Хотя большинство случаев являются спорадическими, описаны и ядерные семьи с аутосомно-доминантным типом наследования синдрома. Клинически синдром ДиДжорджи проявляется задержкой в развитии, «волчьим небом», аплазией или гипоплазией ОЩЖ и тимуса, пороками сердца и характерным внешним видом (квадратный корень носа). Синдром ДиДжорджи – ведущая причина персистирующей гипокальциемии у новорожденных, при этом проявиться гипопаратиреоз у таких пациентов может как в неонатальном периоде, так и позднее в течение жизни. Среди всех людей с этим заболеванием гипопаратиреоз встречается у 60% пациентов.
Синдром HDR (Hypoparathyroidism, Sensorineural Deafness, and Renal Dysplasia Syndrome)
Синдром HDR является следствием мутации в гене GATА3, локализованного в коротком плече 10 хромосомы (10p14–15). Сам GATA3 является транскрипционным фактором, экспрессирующимся у позвоночных в почках, ОЩЖ, тимусе, внутреннем ухе, ЦНС. Описаны случаи как аутосомно-доминантного, так и аутосомно-рецессивного наследования синдрома. Основными проявлениями заболевания являются дисплазия почек, глухота и гипопаратиреоз. Также пациенты с HDR-синдромом чаще всего невысокого роста, что может быть связано с дефицитом соматотропного гормона.
Синдром Саньяд-Сакати и синдром Кенни-Каффи 1 типа
Синдром Саньяд-Сакати и синдром Кенни-Каффи 1 типа характеризуются схожими клиническими проявлениями: врожденным гипопаратиреозом, выраженной задержкой роста, отставанием в умственном развитии, микроцефалией и лицевым дисморфизмом. Синдром Саньяд-Сакати чаще всего встречается у людей с арабскими корнями. У пациентов с синдромом Кенни-Каффи, помимо описанных нарушений, также наблюдаются остеосклероз и иммунодефицит. Оба заболевания являются следствием мутации в гене тубулин-специфичного шаперона Е (TBCE) в локусе, который кодирует белок, отвечающий за связывание тубулина.
Синдром Кенни-Каффи 2 типа
Синдром Кенни-Каффи 2 типа – генетическое заболевание, поражающее преимущественно скелет, глаза, также встречаются различные патологии органов головы и шеи. Одним из частых компонентов заболевания (диагностируется у 30-79% пациентов) является гипопаратиреоз. Заболевание проявляется частыми эпизодами гипокальциемии, а его непосредственной причиной являются различные мутации в гене FAM111А (family with sequence similarity 111 member A). Наследование синдрома аутосомно-доминантное.
Изолированный гипопаратиреоз
Гипопаратиреоз может встречаться изолированно и наследоваться как аутосомно, так и Х-сцепленно.
Аутосомно-рецессивный изолированный гипопаратиреоз. На сегодняшний день в литературе имеется описание нескольких семей с аутосомно-рецессивным гипопаратиреозом. Основной причиной этого заболевания является потеря функции гена GCM2 (glial cells missing), также известного как GCMB в локусе 6p23–24. GCM2 экспрессируется в основном в развивающихся ОЩЖ. Гипопаратиреоз клинически проявляется уже у новорожденных детей вследствие агенезии ОЩЖ. Самыми частыми вариантами мутаций GCM2 являются доминантная ингибирующая или рецессивная инактивирующая. В то же время, гетерозиготные мутации в GCM2 не приводят к каким-либо нарушениям формирования и функционирования ОЩЖ.
X-сцепленный изолированный гипопаратиреоз. Делеции/инсерции в локусах Xq27.1 и 2p25.3 и возникший вследствие этого Х-сцепленный гипопаратиреоз были описаны в двух родственных семьях. У мужчин с этим синдромом с детства возникают гипокальциемические судорожные приступы вследствие изолированного дефекта в развитии ОЩЖ.
Сниженный синтез или секреция ПТГ
Аутосомно-доминантная гипокальциемия (АДГ)
АДГ 1 типа характеризуется активирующей мутацией в гене CASR. CaSR становятся сверхчувствительными к сывороточным концентрациям кальция, а синтез и секреция ПТГ подавляются, несмотря на нормокальциемию. Таким образом, возникает функциональный гипопаратиреоз. АДГ 2 типа возникает при наличии активирующей мутации в гене GNA11 (guanine nucleotide-binding protein alpha 11). GNA11 кодирует альфа-субъединицу G-белка G11, который является ключевым медиатором CaSR-опосредованного сигнального пути. Мутация GNA11 приводит к избыточному подавлению секреции ПТГ даже при гипокальциемии. У пациентов с АДГ развивается умеренная гипокальциемия, тяжелая гипокальциемия может возникнуть в условиях стресса. Почечная реабсорбция кальция при АДГ 1 типа снижена вследствие мутации CaSR в почках; у таких пациентов развивается гиперкальциурия. Почечная реабсорбция кальция при АДГ 2 типа не страдает.
Одним из редких вариантов манифестации АДГ является синдром Барттера с гипокальциемией. Он характеризуется гипокальциемией, гипомагниемией и гипопаратиреозом, а также признаками дисфункции петли Генле (полиурией, гипокалиемическим алкалозом, повышением концентрации ренина и альдостерона в плазме, низким артериальным давлением и резистентностью сосудов к ангиотензину II. Причиной развития синдрома является мутация в локусе 3q21.1, отвечающем за CaSR.
Мутации в гене ПТГ
В литературе описано несколько редких мутаций в гене PTH, нарушающих его синтез и секрецию ПТГ. Например, аутосомно-доминантная мутация в сигнальной последовательности препроПТГ нарушает нормальный процессинг молекулы и, следовательно, образование ПТГ. Также описаны семьи с изолированным гипопаратиреозом, обусловленным аутосомно-рецессивной мутацией в гене PTH.
Антитела к CaSR
При позднем развитии гипопаратиреоза возможной причиной могут являться циркулирующие активирующие антитела к CaSR. Эти антитела не приводят к необратимой деструкции ОЩЖ, поэтому существует вероятность спонтанной ремиссии. В литературе описаны как изолированные формы гипопаратиреоза, вызванного антителами к CaSR, так и случаи, ассоциированные c другими аутоиммунными заболеваниями (тиреоидитом Хашимото, болезнью Аддисона).
Синдромы резистентности к ПТГ
Резистентность органов-мишеней к ПТГ, также известная как псевдогипопаратиреоз (псевдоГПТ), является групповым понятием для нескольких гетерогенных заболеваний, характеризующихся нечувствительностью периферических тканей к ПТГ. По данным лабораторных исследований у таких пациентов обращают на себя внимание гипокальциемия, гиперфосфатемия, повышение уровня ПТГ. Ниже приведено описание основных форм псевдоГПТ.
ПсевдоГПТ 1 типа
ПсевдоГПТ 1 типа является следствием мутации в гене GNAS1, кодирующем альфа-субъединицу стимулирующего G-белка, ассоциированного с рецептором ПТГ. Эти мутации приводят к неспособности G-белка активировать аденилатциклазу после связывания ПТГ с его рецептором, что в итоге приводит к невозможности трансдукции сигнала в органах-мишенях. У людей в гипофизе, гонадах и ЩЖ преимущественно экспрессируется материнский аллель этого гена. В связи с этим клинические проявления заболевания зависят от локализации мутации в материнском или отцовском аллеле. На основании этих различий выделяют несколько подвидов ПсевдоГПТ 1 типа.
ПсевдоГПТ тип 1а. Этот тип псевдоГПТ – аутосомно-доминантное заболевание, вызванное мутацией, приводящей к потере функции GNAS1. Передается псевдоГПТ 1а типа по материнской линии. При этой мутации после связывания ПТГ с рецептором активации аденилатциклазы и трансдукции сигнала не происходит. Клинически псевдоГПТ 1а типа чаще всего проявляется комплексом симптомов, известных как наследственная остеодистрофия Олбрайта: низким ростом, ожирением, короткой шеей, округлым лицом, подкожными кальцинатами, укороченными четвертыми и пятыми метакарпальными и метатарзальными костями и отставанием в развитии. Помимо этого, у пациентов с псевдоГПТ 1а типа может наблюдаться резистентность к другим гормонам, действие которых реализуется через G-белки: гонадотропин-рилизинг гормону, фолликулостимулирующегому гормону (ФСГ), лютеинизирующему гормону (ЛГ) и тиреотропному гормону (ТТГ). В биохимическом анализе крови обращают на себя внимание гипокальциемия с гиперфосфатемией, развивающиеся в результате резистентности почек к действию ПТГ, и вторичный гиперпаратиреоз. Помимо этого, у пациентов может развиваться фиброзный остеит, поскольку резистентность к ПТГ не затрагивает скелет.
Псевдо-псевдоГПТ. При наследовании инактивирующей мутации в гене GNAS1 по отцовской линии также развивается фенотип наследственной остеодистрофии Олбрайта. Однако, при условии наличия нормального материнского аллеля GNAS1 нечувствительность почек к действию ПТГ не возникает. Сывороточные уровни кальция, фосфора и ПТГ у таких пациентов остаются в пределах нормы. Такое состояние получило название псевдо-псевдоГПТ.
Прогрессирующая костная гетероплазия. Унаследованная по отцовской линии инактивация GNAS1 также может вызывать прогрессирующую костную гетероплазию. Данное заболевание дебютирует в раннем детстве и характеризуется эктопическим костеобразованием в дерме, мышцах и соединительной ткани. В клинической картине могут наблюдаться признаки, характерные для наследственной остеодистрофии Олбрайта, не сопровождающиеся изменением концентрация кальция или ПТГ в сыворотке крови.
ПсевдоГПТ типа 1b. ПсевдоГПТ типа 1b – редкое аутосомно-доминантное заболевание, передающееся по материнской линии. Его причиной является мутация, поражающая регуляторные элементы гена GNAS1, а не сам ген. В этом случае резистентность к ПТГ, по всей видимости, развивается только в ткани почек, поэтому у таких пациентов наблюдаются гипокальциемия, гиперфосфатемия и вторичный гиперпаратиреоз без каких-либо проявлений наследственной остеодистрофии Олбрайта.
ПсевдоГПТ типа 1с. Это заболевание является следствием мутации, нарушающей связывание G-белка с рецептором ПТГ. Таким образом, G-белок способен активировать аденилатциклазу, но этот процесс не связан с активацией рецептора ПТГ. Фенотипически такие пациенты сходны с больными с псевдоГПТ 1а типа и имеют все признаки, характерные для остеодистрофии Олбрайта.
ПсевдоГПТ типа 2
У пациентов с псевдоГПТ 2 типа нет клинических проявлений, характерных для наследственной остеодистрофии Олбрайта. Для них характерны повышение концентрации циклического аденозинмонофосфата (цАМФ) в моче, не сопровождающееся гиперфосфатурией в ответ на введение экзогенного ПТГ.
Другие формы резистентности к ПТГ
Резистентность к ПТГ также может быть вызвана рецессивной миссенс-мутацией в последовательности, кодирующей ПТГ (1-84), которая приводит к невозможности связывания гормона с рецептором.
Другие причины гипопаратиреоза
Митохондриальные заболевания
Гипопаратиреоз является компонентом многих митохондриальных заболеваний, возникающих вследствие делеций в митохондриальной ДНК. Ниже приводится краткое описание некоторых митохондриальных заболеваний, сопровождающихся гипопаратиреозом.
Синдром Кернса-Сейра – митохондриальная цитопатия, характеризующаяся энцефалопатией, офтальмоплегией, пигментным ретинитом и кардиомиопатией.
MELAS синдром – сочетание митохондриальной миопатии, энцефалопатии, лактат-ацидоза и инсультоподобных эпизодов. Возникает вследствие точечной мутации в митохондриальной транспортной рибонуклеиновой кислоте (тРНК) и наследуется по материнской линии. Чаще всего манифестирует в детстве, после относительно благополучного периода раннего развития.
Дефицит митохондриального трифункционального белка (MTPDS) – нарушение окисления жирных кислот, манифестирующее некетотической гипогликемией и сопровождающееся кардиомиопатией, дисфункцией печени, скелетной миопатией и задержкой в развитии. В некоторых случаях у матерей детей с MTPDS в период беременности отмечался эпизод острого печеночного повреждения [48,77].
Данные об основных причинах наследственного гипопаратиреоза, ключевых клинических проявлений и лабораторных изменениях представлены в таблице 10.
Таблица 10. Основные причины наследственного гипопаратиреоза [75].
Заболевание | Клинические и лабораторные данные, позволяющие заподозрить заболевание | Молекулярный субстрат заболевания | Необходимые генетические и иные исследования |
|---|
Аутосомно-доминантный гипопаратиреоз 1 и 2 типов | Чаще бессимптомный, реже – с умеренной гипокальциемией, возможна гиперкальциурия | Активирующая мутация CASR (1 тип) или GNA11 (2 тип) | Секвенирование CASR или GNA11 (11 субъединица белка Gα11) |
Аутосомно-доминантный гипопаратиреоз с синдромом Барттера 5 типа | Гипокальциемия, гипомагниемия, гипокалиемия, алкалоз, гиперкальциурия, гипокалиемия и гипонатриемия, обезвоживание в зависимости от тяжести заболевания | CaSR | Секвенирование CASR |
Изолированный гипопаратиреоз | Преобладают клинические и лабораторные симптомы гипопаратиреоза | | PTH, GCM2 секвенирование в зависимости от клинической картины |
Аутосомно-рецессивный | | ПТГ или GCM2 | |
Аутосомно-доминантный | | ПТГ или GCM2 | |
Х-связанный рецессивный | | Локус SOX3 (у мужчин) | |
HDR-синдром (гипопаратиреоз, глухота, дисплазия почек) | Сенсоневральная тугоухость, анатомические особенности развития почек и нарушения их функционирования, аутосомно-доминантный тип наследования | Ген GATA3 | Секвенирование гена GATA3, слуховые тесты, визуализация почек |
Синдром ДиДжорджи | Аномалии сердечно-сосудистой системы, в т.ч. тетрада Фалло, дефекты межжелудочковой перегородки, truncus arteriosus, патологии дуги аорты (встречаются у ~80%); иммунодефицит (частые инфекционные заболевания, а- или гипоплазия тимуса, Т-клеточная лимфопения), гипопаратиреоз, аномалии строения глотки и гортани, волчья пасть, поведенческие и психиатрические нарушения, офтальмологические нарушения, потеря слуха | Различные дефекты, в т.ч. делеции и микроделеции в 22q11.2 хромосоме | |
Синдром Кенни-Каффи |
Тип 1 или синдром Саньяд–Сакати (аутосомно-рецессивный) | Низкий рост, задержка роста, маленький размер кистей и стоп, кортикальное истощение и медуллярный стеноз трубчатых костей, отложенное закрытие родничков, дисморфизм лица, аномалии глаз, гипопаратиреоз | Ген TCBE | Секвенирование гена TCBE |
Тип 2 (аутосомно-доминантный) | Дисплазия костей, низкий рост, кортикальное истончение и медуллярный стеноз трубчатых костей, отложенное закрытие родничков, дисморфизм лица, аномалии глаз, гипопаратиреоз | Ген FAM111A | Секвенирование гена FAM111A |
Гипопаратиреоз, ассоциированный с митохондриальными болезнями |
| Синдром Кернса-Сейра | Офтальмоплегия, нарушения пигментации сетчатки и внутрисердечной проводимости, проксимальная и бульбарная слабость, возможна атаксия | Крупная делеция митохондриальной ДНК | Специальные клинические обследования, в зависимости от поражения органов и систем |
| Синдром MELAS | Энцефаломиопатия, лактат-ацидоз, инсультоподобные эпизоды, офтальмопления, диабет, потеря слуха, мигрени и когнитивные нарушения | Мутация в митохондриальном гене тРНК | |
| Синдром MTPDS | Нарушения в окислении жирных кислот, ассоциированные с нейропатией, ретинопатией и ожирением печени | Мутации в митохондриальном геноме | |
GNA11 – G protein alpha subunit 11, GATA3 – GATA-binding protein 3, FAM111A – family with sequence similarity 111 member A, GCMB – glial cell missing gene, HRD – hypoparathyroidism retardation dismorfism, SOX3 – sry-related HMG box, TBCE – tubulin folding cofactor E, MTPDS – mitochondrial trifunctional protein deficiency