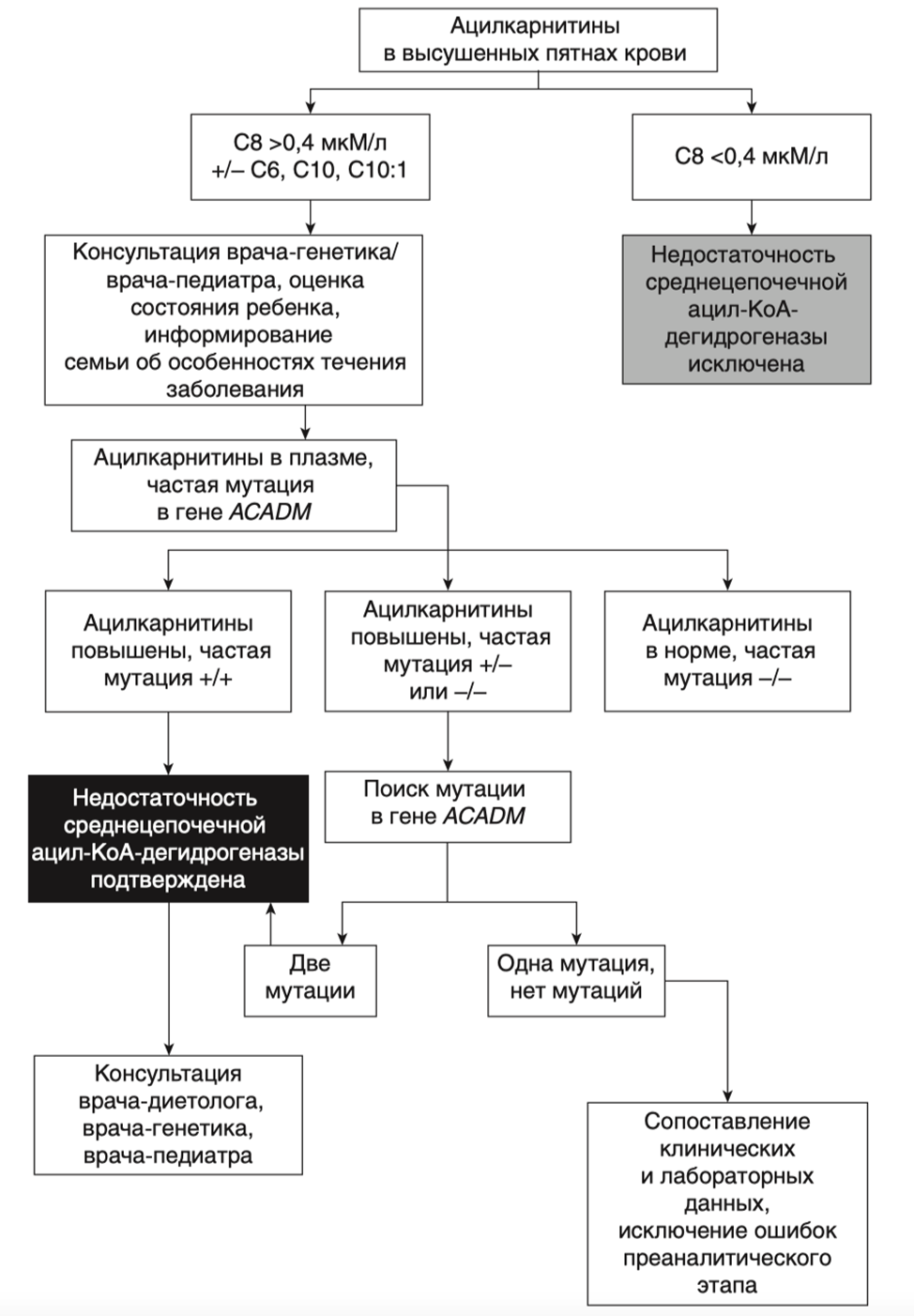
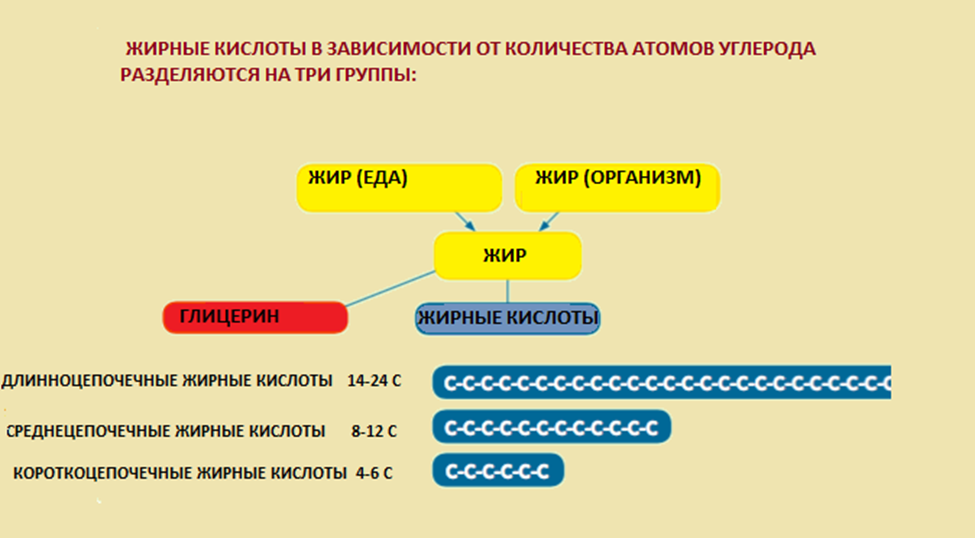
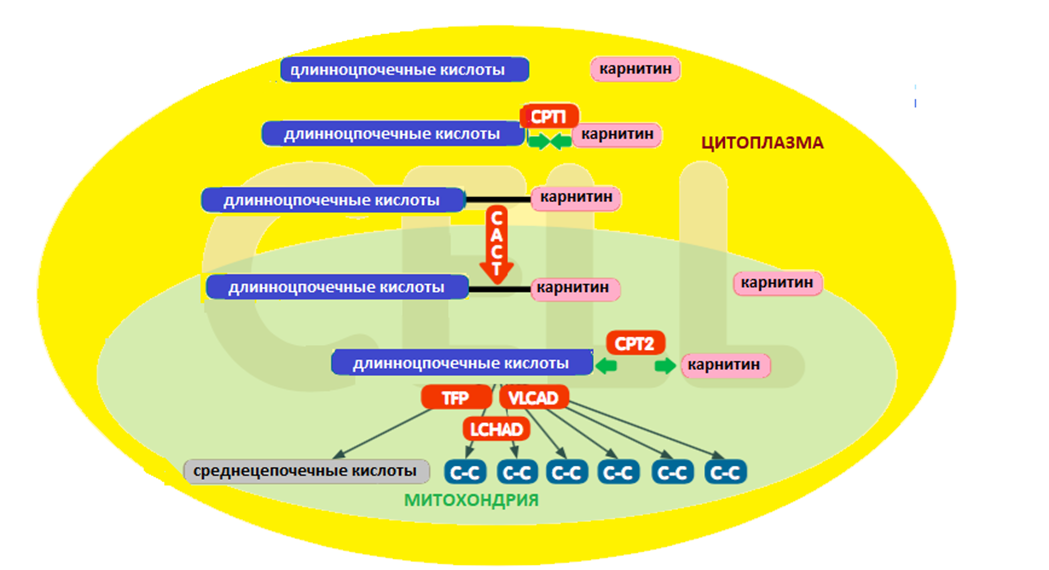
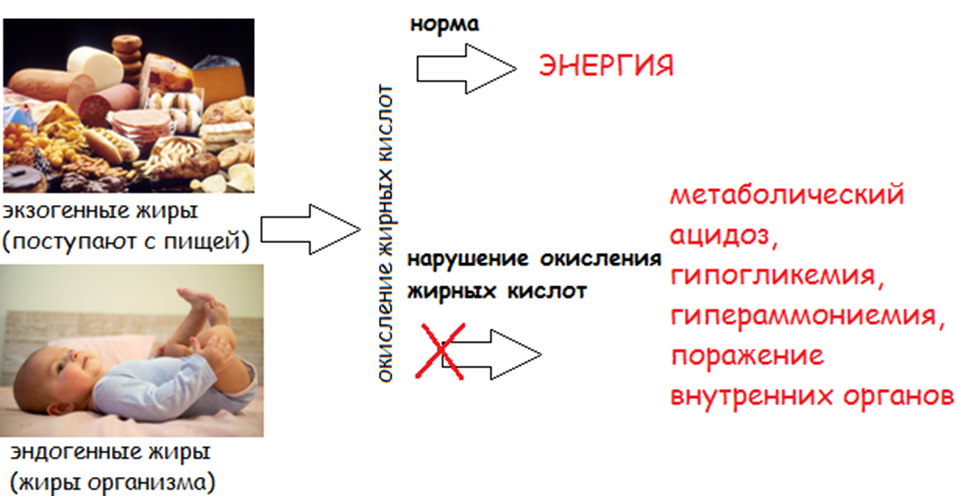
3.1 Общие рекомендации по диетотерапии пациентов с FAOD
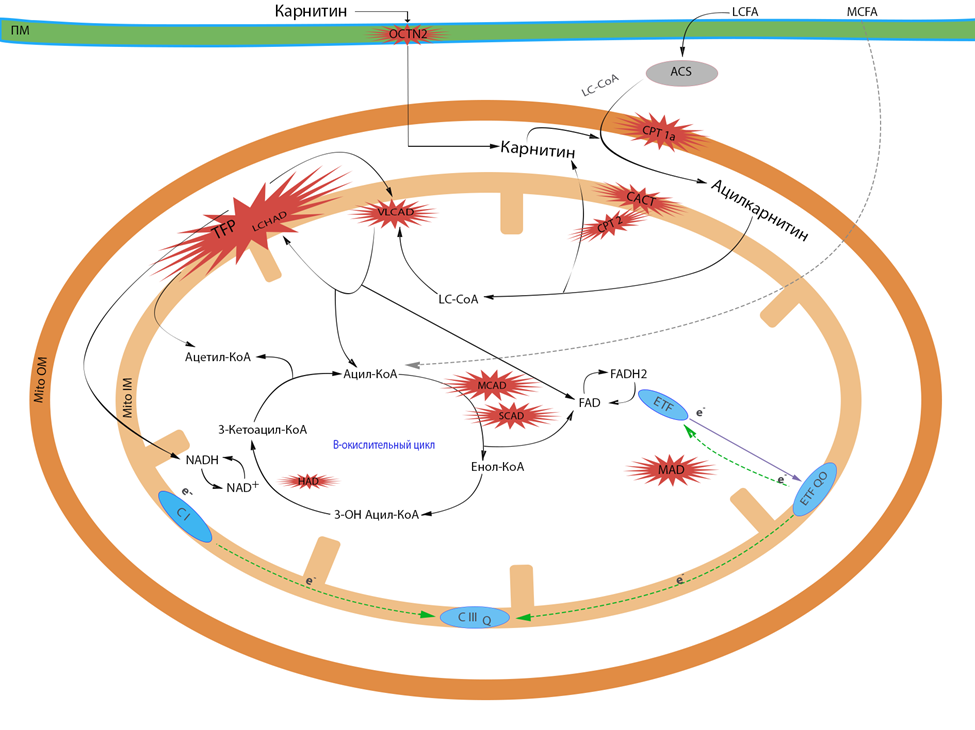
Стратегия лечения пациентов с FAOD носит комплексный характер, в его основе лежит коррекция метаболических нарушений посредством диеты, которая заключается в снижении потребления пищевых жиров в качестве резервной составляющей тканевой биоэнергетики, минимизации катаболизма жирных кислот и уменьшении их значимости для восполнения энергозатрат клетки с обеспечением нормальных процессов анаболизма, роста и нутритивного статуса. Главная задача диетотерапии – это профилактика голодания, предупреждение гипогликемии и минимально допустимое снижение поступления с пищей патогенетически значимых жирных кислот и их источников.
Поступление достаточной энергии за счет углеводов является ключевым моментом в диетотерапии FAOD. Процесс анаболизма подавляет окисление жирных кислот и снижает уровень циркулирующих ацилкарнитинов. Расчет калорийности питания должен обеспечивать адекватное поступление энергии, препятствуя катаболизму, и в то же время не приводить к перекармливанию и к патологической прибавке веса. По результатам проведенных исследований у пациентов с FAOD были отмечены следующие особенности метаболизма [81]:
- более низкие энергозатраты;
- преимущественное окисление углеводов для получения энергии;
- низкая мышечная масса тела и большая жировая масса тела.
- Рекомендуется избегать длительного голодания всем пациентам с FAOD для предотвращения развития метаболических кризов [11-14, 145].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарий: в приложении А8 суммированы рекомендации по продолжительности периодов голодания в соответствии с возрастом. Однако указанные значения применимы к здоровым детям и стабильным состояниям, когда в условиях нормокалорийного питания дневные запасы гликогена используются в период продолжительного ночного голодания для продукции глюкозы. Необходимо учитывать индивидуальные особенности ребенка, для некоторых пациентов требуются меньшие интервалы между приемами пищи для сохранения стабильного состояния.
Начиная с 8–12 месяцев, с целью предотвращения катаболизма в ночной период, можно давать перед сном кукурузный крахмал (1 г/ кг /день (разведение крахмала в детской смеси или в воде 1:2), при регистрации гипогликемии в ночные часы возможно дополнительное введение крахмала в той же дозе) для обеспечения поступления адекватной энергии в течение ночи, данная тактика обычно используется только у пациентов с тяжелым течением заболевания, склонных к гипогликемии. В приложении А9 приведены среднесуточные нормы физиологических потребностей в энергии для детей.
- Рекомендовано назначение диетотерапии всем пациентам с FAOD (кроме MCADD, SCADD, системной недостаточности карнитина) после установления точного диагноза с целью снижения рисков возникновения метаболической декомпенсации [11-14].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 4).
Комментарии: в зависимости от формы заболевания рекомендуются разные схемы лечения. В Приложении А10 приведены основные рекомендации по соотношению жиров и углеводов в диете.
3.1.1 Лечение пациентов с FAOD в период интеркуррентных инфекций
У пациентов с FAOD эпизоды метаболической декомпенсации могут наблюдаться при интеркуррентных заболеваниях, гастроэнтерите.
- Рекомендуется поддерживать высокое потребление углеводов во время любого метаболического стресса всем пациентам с FAOD для предотвращения развития метаболического криза [110].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5)
Комментарии: напитки с 20–25% раствором декстрозы или кукурузного крахмала (кукурузный крахмал - пациентам старше 8 месяцев жизни в разовой дозе 1 г/кг) следует начинать при первых признаках заболевания, а затем равномерно распределять в течение суток. Для тех пациентов, у которых установлен зонд проводится непрерывное энтеральное питание или повторный болюс питательного раствора, уже используемого ночью, может быть предложен в течение всего дня. В случаях клинического ухудшения с отказом от еды, рвотой, необходима госпитализация для проведения инфузионной терапии (Приложение Г 9).
3.1.2 Рекомендации по лечению пациентов с FAOD в период метаболической декомпенсации
У всех пациентов с FAOD, независимо от наличия или отсутствия клинических симптомов, катаболические состояния требуют применения режима неотложной ситуации.
Состояние метаболического криза является показанием для госпитализации и проведения интенсивной, в том числе инфузионной терапии, которая должна начинаться незамедлительно. Тактика лечения детей в период криза включает дополнительное введение декстрозы** для энергетической поддержки и уменьшения интенсивности процессов катаболизма, коррекцию метаболического ацидоза, гипераммониемии и водно- электролитных нарушений, коррекцию диетотерапии.
Устранение гипогликемии и энергетической недостаточности имеет первостепенное значение для сохранения жизни и здоровья пациентов с FAOD.
- Рекомендуется внутривенное введение раствора декстрозы** под контролем ее уровня в крови пациентам с FAOD при развитии метаболического криза [5, 12 ,52, 130, 150].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 4).
Комментарии: Внутривенное введение раствора декстрозы** из расчета: новорожденные и дети до 3-х лет 10-12 мг/кг/мин, от 3 до 10 лет 8-10 мг/кг/мин, старше 10 лет 5-8 мг/кг/мин. При снижении глюкозы ниже 2,5 ммоль/л в крови необходимо внутривенное струйное введение не менее 10% декстрозы** из расчета 2-3 мл/кг, далее переходят на внутривенное капельное введение 10% декстрозы** со скоростью 7 - 9 мг/кг/мин до нормализации уровня глюкозы в крови.) Назначение декстрозы** не только восполняет тканевой энергетический дефицит, но и подавляет липолиз и снижает продукцию токсичных дериватов жирных кислот.
Неотложную внутривенную терапию следует начинать как можно раньше при более тяжелых состояниях, с высокой температурой, повторяющейся рвотой или тяжелым гастроэнтеритом.
Растворы для внутривенных инфузий, содержащие декстрозу** следует использовать даже при нормальном уровне глюкозы в крови. Пациенты с FAOD полностью зависят от глюкозы для своих энергетических потребностей и ее запас может быстро истощаться. Если у пациента развивается значительная гипергликемия (например, уровень глюкозы в крови больше 13,8 ммоль/л) с глюкозурией и имеются признаки метаболической декомпенсации, то следует рассмотреть возможность начала инфузии #инсулин растворимый (человеческий генно-инженерный) ** вместо снижения скорости инфузии декстрозы**, способствуя анаболическому состоянию. Начальная доза для инфузии #инсулина растворимого (человеческий генно-инженерный)** составляет 0,01 МЕ/кг/мин. Уровень инсулина следует титровать, чтобы поддерживать уровень глюкозы в крови между 5,5 и 8,3 ммоль/л [153, 160].
Следует отметить, что инфузия более концентрированного раствора декстрозы** при более низкой скорости инфузии посредством центрального катетера может потребоваться при недостаточной сердечной функции. Кроме того, следует соблюдать осторожность при прекращении внутривенной инфузии - необходимо медленно снижать скорость, чтобы избежать состояния реактивной гипогликемии.
- Рекомендуется коррекция метаболического ацидоза (при уровне бикарбонатов сыворотки крови <16 мЭкв/л) пациентам с FAOD при развитии метаболического криза [5, 12, 52].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 4).
Комментарии: согласно клиническим рекомендациям по интенсивной терапии дефицит бикарбонатов купируется путем внутривенного введения щелочных растворов: натрия гидрокарбонат**. Натрия гидрокарбонат** применяется в виде 8,4% и 4,2% раствора для удобства перерасчета на ммоль NаНСО. Его дозировка (ммоль) определяется по формуле: (-ВЕ) Х масса тела (кг) Х 0,3. Также пациентам может быть назначено щелочное питье – раствор соды из расчета 1⁄2-1 чайная ложка на 200 мл воды, щелочные минеральные воды.
В зависимости от тяжести состояния каждые 6-12 часов контролировать показатели кислотно-основного состояния крови, уровня натрия и калия в крови пациентам с FAOD при развитии метаболического криза [5,12,13,14].
- Рекомендуется коррекция водно-электролитных нарушений пациентам с FAOD при развитии метаболического криза [5,12, 145].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: при сохраняющейся гипогидратации проводится путем внутривенного введения натрия хлорида** 0,9%). Однако необходимо иметь в виду, что главным мероприятием в комплексе интенсивной терапии является введение растворов декстрозы** и щелочных растворов.
- Рекомендовано: назначение натрия бензоата (биологически активная добавка) при уровне аммиака в крови выше 150-200 мкмоль/л пациентам с FAOD при развитии метаболического криза [152, 154].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: Натрия бензоат (биологически активная добавка) назначается из расчета от 250 мг\кг \в сутки. (максимально до 500мг/кг/сут, если масса тела превышает 20 кг до 5,5г/м2/сут)
Исследование уровня аммиака в крови необходимо проводить каждые 6-12 часов. Осуществляют исследование уровня натрия и калия в крови не реже 1раза в 2 часа.
Согласно международным рекомендациям по лечению гипераммониемии показано внутривенное введение препаратов, связывающих аммиак, однако в настоящее время на территории Российской Федерации данные лекарственные средства не зарегистрированы. Поэтому все препараты, связывающие аммиак, являются препаратами off-label или не зарегистрированы и требуют оформления врачебной комиссии и согласия родителей/законных представителей и пациента с 15 лет. Прием данных препаратов при гипераммониемии необходим по жизненным показаниям
- Рекомендуется коррекция диетических мероприятий всем пациентам с FAOD в период метаболического криза с целью компенсации данного состояния [1, 105].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: при VLCAD, LCHAD диетические ограничения при кризе: полностью исключить потребление натуральных (ДЦТ) жиров на период острогоa криза (на 24-48 часов). Суточная потребность в жирах обеспечивается только за счет СЦТ. После 24-48 часов - вводить натуральные жиры до достижения рекомендуемой возрастной потребности, под контролем лабораторных показателей и состояния пациента. Энергетическая ценность рациона в период метаболическогно криза должна быть увеличена минимум на 10% от суточной возрастной нормы, в основном за счет углеводов; избегать голодания, когда прекращаются инфузии.
- Рекомендуется использовать приемлемый энтеральный способ кормления для пациента с FAOD для предотвращения развития катаболических состояний [1, 105].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: самостоятельно через рот, через зонд или гастростому.
Кормление через назогастральный зонд в рамках долгосрочной терапии, в целом, нежелательно, но рекомендуется в условиях заболевания на начальном этапе метаболического нарушения. В повседневной практике назначение декстрозы** или кукурузного крахмала через рот не требуется, а применяется только при угрозе возникновения или во время метаболического криза.
- В случае необходимости введения эпинефрина** всем пациентам с FAOD рекомендовано его введение вместе с 10% раствором декстрозы** [97, 151].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: Эпинефрин** может стимулировать липолиз, поэтому при назначении этим детям следует добавлять декстрозу**
Международные клинические рекомендации по лечению разработаны только для отдельных форм FAOD – дефектах среднецепочечной дегидрогеназы жирных кислот, очень длинноцепочечной ацил-КоА дегидрогеназы жирных кислот и длинноцепочечной 3-ОН ацил-КоА дегидрогеназы жирных кислот. Для других форм FAOD рекомендации по диетотерапии могут различаться в разных странах. Ниже приводятся основные рекомендации, которые по мнению рабочей группы являются наиболее полными.
3.2 Рекомендации по терапии отдельных форм FAOD
В зависимости от первичного метаболического дефекта подходы к терапии могут существенно различаться или иметь минорные различия.
3.2.1 Рекомендации по лечению системной недостаточности карнитина
- Не рекомендовано назначение специальной диетотерапии пациентам с системной недостаточностью карнитина, поскольку ведущим патогенетическим механизмом развития заболевания является дефицит карнитина [82].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: восполнение карнитина является основным методом лечения данной патологии. Также пациентам следует избегать перерывов в приеме пищи (см. общие рекомендации для FAOD и Приложение А7).
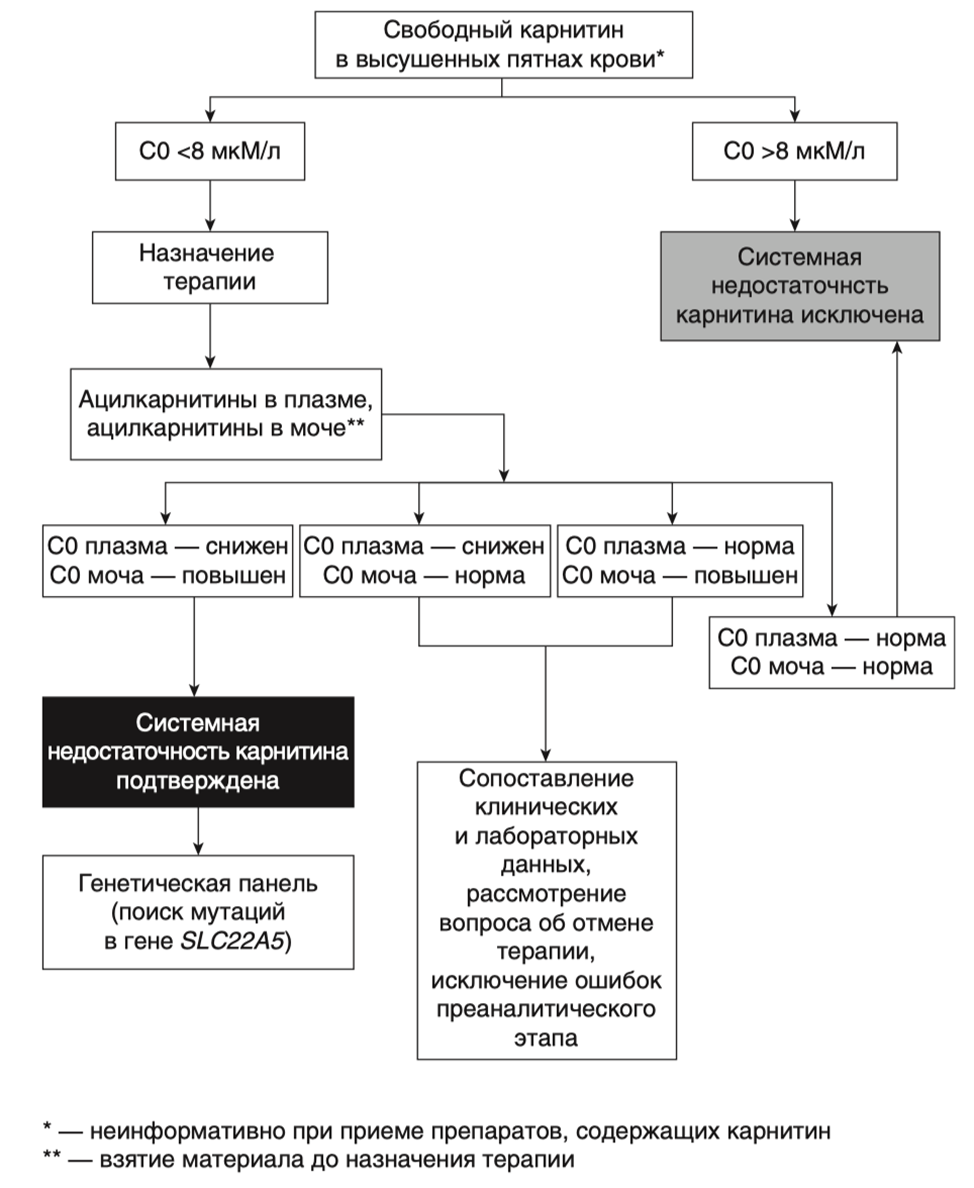
- Рекомендовано назначение #левокарнитина в дозе 50–400 мг/кг/сутки, всем пациентам с подтвержденной первичной недостаточностью карнитина с целью снижения риска возникновения гипогликемических эпизодов [54, 82, 155].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: #Левокарнитин назначают в дозе 50–400 мг / кг / сутки, разделенной на три приема. Точная доза #левокарнитина должна быть скорректирована в зависимости от уровня карнитина в плазме. Концентрацию карнитина следует измерять как минимум через 4 ч после последнего приема препарата. Концентрация свободного карнитина у пациентов с первичным дефицитом карнитина сохраняется ниже нормы даже при его применении [14]. Не существует установленного «терапевтического» уровня, которого следует достигнуть и адекватной считается концентрация свободного карнитина> 20 мкМ. Уровень карнитина в плазме быстро падает после прекращения терапии #Левокарнитином и затем происходит истощение его запаса в тканях. Это объясняет, почему пациенты с первичным дефицитом карнитина, которые пропускают прием препарата, остаются бессимптомными несколько месяцев, что может привести к тому, что они прекращают прием #левокарнитина. Эти пациенты подвергаются риску внезапной смерти из-за остановки сердечной деятельности. Очень важно продолжать терапию ежедневно, чтобы избежать тяжелых нарушений со стороны сердца. Длительная терапия #Левокарнитином полностью купирует клинические проявления заболевания с нормализацией размеров сердца и ЭКГ в течение месяца с восстановлением мышечного тонуса, хотя тканевые (в мышечной ткани) концентрации карнитина остаются очень низкими (<10 % от N) в течение длительного времени.
Прием #Левокарнитином в высоких дозах может вызывать усиление моторики желудочно-кишечного тракта, диарею, и выработку триметиламина, что является причиной «рыбного запаха» от мочи и тела. Уменьшение дозы карнитина может уменьшить эти побочные эффекты. Если неприятный запах сохраняется, возможно назначение курса перорального приема #метронидазола** в дозе 10 мг / кг / сутки в течение 7–10 дней [82, 83, 155, 156].
3.2.2 Рекомендации по лечению дефицита карнитин-ацилкарнитин транслоказы
Клинические рекомендации, основанные на достаточном уровне доказательности и убедительности, для лечения и мониторинга этого конкретного заболевания в мире не разработаны. Были опубликованы только рекомендации, основанные на опыте экспертов по клиническому и биохимическому мониторингу пациентов с FAOD. Нет единого мнения относительно использования левокарнитина при этой форме болезни. Некоторые авторы не рекомендуют этого, поскольку накопление длинноцепочечных ацилкарнитинов может быть токсичным для сердечного ритма [84]. Тем не менее, другие авторы считают целесообразным применять его при эпизодах метаболической декомпенсации, для восстановления митохондриального пула коэнзима А [66, 85-89].
- Рекомендуется пациентам с установленным диагнозом СAСT соблюдение низкожировой диеты (потребление жиров должно составлять 30% или менее от общей калорийности, 20% из них - MCT с низким содержанием C10 и 10% LCT (длинноцепочечные триглицериды) для предотвращения развития метаболического криза [66, 87-89].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: рекомендовано MCT с низким содержанием C10 в связи с тем, что жиры C10 требуют для окисления активной работы CACT [90].
- Рекомендуется пациентам с установленным диагнозом СAСT избегать длительных перерывов в приеме пищи для предотвращения гипогликемических состояний [66, 87-89].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: не существует единого мнения о безопасных интервалах между кормлениями, в данных клинических рекомендациях нами были выбраны те, которые опубликованы в работе группы экспертов из европейских стран. Максимально допустимый безопасный интервал для разных возрастных групп приведен в Приложении А9 [66].
- Рекомендуются пациентам с установленным диагнозом СAСT добавки незаменимых жирных кислот, с целью предотвращения их дефицита - линолевой (3–4%) и линоленовой (0,5–1%) - в соотношении от 5:1 до 10:1, их источником могут являются масло грецкого ореха, льняное, соевое [66, 87-89].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5)
Комментарии: Содержание кислот в разных маслах приведено в Приложении А16 [66].
- Рекомендуется пациентам с установленным диагнозом СAСT в период интекуррентных инфекций и других состояний, при которых активируются механизмы катаболизма, применение перорально растворов декстрозы** или полимеров декстрозы в виде пищевой добавки (мальтодекстрин (декстринмальтоза)) и повышенное потребление углеводов [89].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
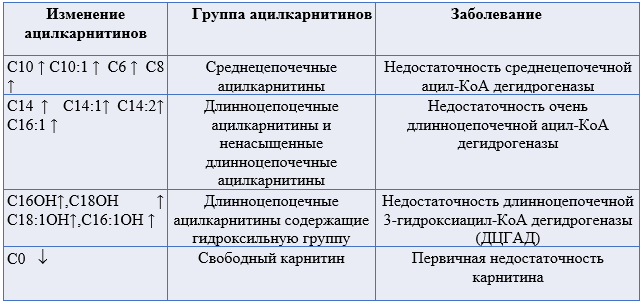
Комментарии: детальные рекомендации см. в разделе 3.1.2 Рекомендации по лечению пациентов с FAOD. В период метаболической декомпенсации снижение уровня длинноцепочечных ацилкарнитинов считается важным маркером эффективности лечения.
3.2.3 Рекомендации по рациону питания при дефиците карнитин пальмитоил-CoA трансферазы I, II
Клинические рекомендации, основанные на достаточном уровне доказательности и убедительности, для лечения и мониторинга дефицита CPT2 и CPT1 в мире не разработаны. Были опубликованы только рекомендации, основанные на опыте экспертов по клиническому и биохимическому мониторингу пациентов с FAOD.
- Рекомендуется пациентам с установленным диагнозом CPT 2 и CPT 1 избегать длительных перерывов в приеме пищи для предотвращения гипогликемических состояний. [66].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: частое кормление каждые 3-4 часа рекомендуется, особенно для детей, учитывая их ограниченные запасы гликогена. Добавление кукурузного крахмала на ночь и в ночные кормления определяется уровнем гликемии и обеспечивает постоянный источник углеводов с медленным высвобождением для предотвращения гипогликемии во время сна. Дети более старшего возраста не должны иметь ночные перерывы в приеме пищи более 12 часов, а в период инфекционного заболевания перерывы в приеме пищи должны быть еще меньше. Взрослые пациенты должны быть проинформированы о рисках, связанных с голоданием, и они, и их лечащий врач должны знать о рисках во время хирургических операций и анестезии. Следует рекомендовать внутривенное введение декстрозы**, если необходимо голодать более 12 часов из-за болезни или медицинских процедур. Максимально допустимый безопасный интервал для разных возрастных групп приведен в Приложение А8
- Рекомендуется диета с высоким содержанием углеводов (70% калорий) с низким содержанием жира (<20% калорий) пациентам с установленным диагнозом дефицит CPT 2 и CPT 1 для предотвращения гипогликемических состояний и метаболической декомпенсации [91].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
- Рекомендуется в период интеркуррентных инфекций и других состояний, при которых активируются механизмы катаболизма, пациентам с установленным диагнозом дефицит CPT 2 и CPT 1 с целью предотвращения метаболической декомпенсации употреблять примерно треть от общего количества калорий в виде МСТ [91].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: жирные кислоты C6-C10 не требуют карнитинового челнока для входа в митохондрию, поэтому МСТ может применяться в качестве основного источника энергии.
- С целью профилактики эпизодов миоглобинурии пациентам с установленным диагнозом CPT 1, CPT2 рекомендуется избегать физических нагрузок во время интеркуррентных инфекций и при длительных перерывах в приеме пищи [91, 92].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
- Взрослым пациентам с установленным диагнозом CPT 1, CPT2 в период метаболического криза, сопровождаемого рабдомиолизом и миоглобинурией рекомендуется применение стандартного протокола лечения этого осложнения, чтобы предотвратить острую почечную недостаточность [13, 92, 93].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: при приступе миоглобинурии проводят внутривенное введение 0,9% раствора натрия хлорида** и натрия гидрокарбоната ** (натрия хлорида** 110 ммоль / л, натрия гидрокарбоната** 40 ммоль / л) в 5% растворе декстрозы**, к которому добавляют 10 г #маннитола** на литр (из 10% или 15% раствора). Раствор следует вводить молодому взрослому человеку весом 75 кг из расчета 12 л/сут, чтобы получить диурез 8 л/сут и поддерживать рН выше 6,5. Этот терапевтический режим позволяет контролировать гиперкалиемию и ацидоз и, следовательно, может предотвратить острую почечную недостаточность. Если пациенты не реагируют на регидратацию, может потребоваться гемодиализ. Гемодиализ корректирует метаболический ацидоз и нарушения электролитов и способствует удалению мионекротических токсинов из плазмы. Диализ показан при почечной недостаточности, если у пациента анурия и диурез не восстановлен после регидратации. Есть основания полагать, что упреждающее начало гемодиализа может улучшить исходы путем удаления нефротоксинов и предотвращения угрожающих жизни осложнений, таких как гиперкалиемия и глубокий метаболический ацидоз. Лечение острой почечной недостаточности следует проводить в соответствии с клиническими рекомендациями, разработанным профессиональными сообществами по нефрологии.
3.2.4 Рекомендации по рациону питания при дефиците SCAD
Так как большинство пациентов с SCADD не имеют симптомов, лечение не требуется, не существует общепринятых рекомендаций по диетотерапии или применению левокарнитина и рибофлавина (биологическая активная добавка).
3.2.5 Рекомендации по рациону питания при дефиците MCAD
Во многих странах проводится неонатальный скрининг на недостаточность MCAD, поэтому пациентов выявляют до начала клинических симптомов. Рекомендации по диетотерапии различаются в зависимости от возраста и статуса подтверждения диагноза.
- Рекомендуется пациентам с установленным диагнозом на недостаточность MCAD прием левокарнитина в дозе 60-100 мг на кг в сутки для коррекции вторичного дефицита с целью усиления выведения токсичных метаболитов [130, 149].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: Профессиональное сообщество не выработало единых рекомендаций по применению Левокарнитина при MCAD. Некоторые авторы рекомендуют 100 мг / кг / день левокарнитина, чтобы компенсировать вторичный дефицит карнитина и улучшить выведение токсичных метаболитов, другие считают, что в этом нет необходимости [94]. В исследованиях переносимости физических нагрузок у пациентов с дефицитом MCAD было показано, после приема Левокарнитина 100 мг / кг / день переносимость физической нагрузки возрастает [95]. Хотя у пациентов с дефицитом MCAD не было отмечено серьезных побочных эффектов при приеме Левокарнитина, некоторые пациенты жаловались на тошноту, диарею, боль в животе и рыбный запах при приеме в дозе 100 мг / кг / день [96]. Учитывая эту информацию, некоторые врачи рекомендуют использовать низкие дозы Левокарнитина, если уровень свободного карнитина в крови ниже нормы.
3.2.5.1 Рекомендации по диетотерапии новорожденных с MCAD, выявленных при неонатальном скрининге
- Рекомендуются соблюдать интервалы между кормлениями не более 3-4 часов для доношенных детей с выявленными изменениями в анализе крови при проведении неонатального скрининга, характерными для дефицита MCAD, пока диагноз не будет подтвержден или исключен на основании дополнительных тестов [98].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: Считается, что новорожденные при дефиците MCAD подвергаются наибольшему риску развития гипогликемии в течение первых 72 часов жизни, особенно при грудном вскармливании. Необходимо соблюдать регулярные кормления, не допустимы пропуски ночных кормлений!
- Рекомендуется соблюдать «безопасные» интервалы между кормлением для новорожденных с установленным диагнозом MCAD для предотвращения эпизодов метаболической декомпенсации (Приложение А9) [98].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: безопасные интервалы между кормлениями были определены на основании опыта стран, проводящих скрининг на дефицит MCAD.
- Рекомендуется новорожденным с установленным диагнозом недостаточность MCAD при грудном вскармливании дополнить питание адаптированной молочной смесью в течение первых трех полных дней (72 часа) для достижения необходимой потребности в калориях [98].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: Дети с MCAD, вскармливаемые грудным молоком, подвергаются особому риску, поскольку содержание энергии в грудном молоке в течение первых нескольких дней низкое, и его объёмы небольшие. Поэтому рекомендуется дополнить питание адаптированными молочными смесями 60 мл / кг / день. Желательно равномерно разделить объем смеси между 6 и 8 кормлениями и давать после кормления грудью. Если ребенок не усваивает достаточный объем смеси и грудного молока, следует назначить кормление через назогастральный зонд или назначить внутривенную инфузию 10% декстрозой** для достижения необходимой потребности в калориях.
3.2.5.2 Рекомендации по диетотерапии пациентов с MCAD
- Рекомендуется всем пациентам с установленным диагнозом недостаточность MCAD соблюдать интервалы между кормлениями для предотвращения развития гипогликемических состояний (Приложение А8) [98, 94].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: Режим питания у детей старшего возраста и взрослых пациентов должен быть регулярным и включать три основных приема пищи (завтрак, обед, ужин), а также 2-3 перекуса (2-ой завтрак, полдник и прием высокоуглеводного продукта перед сном (хлебобулочные изделия, галеты, крупяное блюдо) с напитком (кисель, компот, чай). Интервал голодания в ночные часы не должен превышать 12 часов.
- Рекомендуется младенцам первого года жизни с установленным диагнозом недостаточность MCAD кормление материнским молоком или молочными смесями без ограничения LCF и без добавок МСТ. Рекомендуется соблюдать интервалы между кормлениями для предотвращения развития гипогликемических состояний (Приложение А8) [98].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: небольшие количества МСТ присутствуют в обычных детских смесях, но их количества минимально и не может быть опасным. В высокой концентрации MCT добавляют в некоторые специальные молочные смеси и их не рекомендуют детям с данной формой болезни. Необходимо отказаться от специальных диетических продуктов, содержащих в больших количествах МСТ.
Жиры в рационе у пациентов с недостаточностью MCAD не ограничиваются [98].
Среднецепочечные жиры встречаются в некоторых продуктах, например, в сливочном масле, коровьем молоке в небольших количествах, и они могут быть включены в рацион без особых ограничений. Кокос является единственным исключением, где 5% жирных кислот имеют длину цепи С8 или С10. Рекомендуют избегать употребления чистого кокоса и кокосового масла, поскольку неизвестно влияние их большого количества на состояние здоровья пациентов с недостаточностью MCAD. MCT добавляются к некоторым специальным смесям для детей и энергетическим добавкам, и их не следует включать в рацион питания пациентов. Важно проверить текущий состав всех продуктов на предмет присутствия MCT.
- Рекомендовано у взрослых пациентов при дефиците MCAD избегать длительного голодания для предотвращения гипогликемических состояний и метаболических кризов [94, 98].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
- Рекомендовано при наличии у пациента с недостаточностью MCAD признаков рекуррентного заболевания, выпаивание растворами, содержащими полимер декстрозы, чтобы предотвратить метаболическую декомпенсацию [94, 98].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: Рекомендуют также в период болезни исключать жиры, хотя убедительных доказательств необходимости в этом для недостаточности MCAD не получены. По мере улучшения состояния ребенка нормальная диета может быть возобновлена, но следует давать дополнительные «энергетические» напитки, особенно ночью, до тех пор, пока ребенок полностью не выздоровеет и не перейдет на обычный режим питания.
- Рекомендовано при наличии у пациента с недостаточностью MCAD в период метаболической декомпенсации в случае сложностей приема растворов через рот проведение внутривенной инфузионной терапии растворами, содержащим декстрозу** [94, 98].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: Следует начинать инфузионную терапию растворами декстрозы** при одном из следующих показаний: многократная рвота, гипогликемия, низкий PO2, обезвоживание, нарушения сознания, метаболический ацидоз. Подробно описано в разделе 3.1.2 Рекомендации по лечению пациентов с FAOD в период метаболической декомпенсации
- Не рекомендовано пациентам с недостаточностью MCAD в период метаболической декомпенсации введение жиров с целью предотвращения усугубления тяжести состояния [98, 99].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
- Рекомендуется всем взрослым пациентам с установленным диагнозом недостаточность MCAD избегать диеты с высоким содержанием жиров, употребление алкоголя, что вызывает метаболическую декомпенсацию [94, 99].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
- Рекомендуется избегать применения ацетилсалициловой кислоты** всем пациентам с установленным диагнозом MCAD с целью предотвращения развития метаболического криза и синдрома Рейе [94, 100].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: Ацетилсалициловая кислота** увеличивает митохондриальное окисление жирных кислот, что способствует их накоплению, а также ингибирует пероксисомное окисление.Это приводит к ухудшению состояния пациентов, поскольку окисление жиров в пероксисомах является альтернативным метаболическим путем, позволяющем частично компенсировать биохимический дефект при FAOD.
3.2.6 Рекомендации по рациону питания при дефиците LCHAD и TFP
Дефицит LCHAD - одна из самых тяжелых форм FAOD, при которой наиболее часто развиваются метаболические кризы. Соблюдение диеты рекомендовано всем пациентам в раннем детском возрасте вне зависимости от наличия или отсутствия клинических симптомов [101].
- Рекомендовано у пациентов при дефиците LCHAD и TFP придерживаться максимально низкого потребление ДЦТ, как при наличии симптомов, так и при их отсутствии, для профилактики возникновения полинейропатии [66].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: Новорожденным показан прием специальной смеси с низким содержанием ДЦТ и высоким содержанием MCT (MCT-содержащая смесь). MCT-содержащая специальная смесь обеспечивает все потребности в питательных веществах. Однако в этом случае необходим дополнительный прием незаменимых длинноцепочечных жирных кислот.
- Рекомендуется назначение диетотерапии при выявлении характерных изменений по результатам неонатального скрининга и/или биохимической диагностики, не дожидаясь подтверждения диагноза молекулярно-генетическим методом [66].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: Поскольку у младенцев может наблюдаться активный липолиз, концентрация некоторых ацилкарнитинов может превышать нормальные значения, что является причиной ложноположительных результатов анализа. В случае если диагноз не будет подтвержден, диетотерапия может быть отменена.
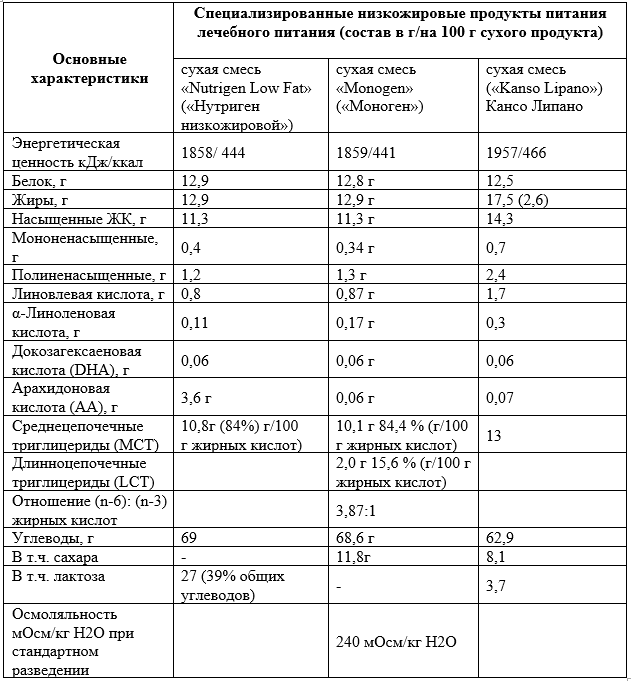
- Рекомендуется при введении твердой пищи в рацион пациентов с недостаточностью LCHAD и TFP поддерживать содержание общих жиров в рационе 25–30% от общего потребления калорий, где 20–25% приходится на MCT и 5–10% - на LCT (Приложение А14) [66].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: Описан положительный эффект повышения потребления белка в отношении энергетического баланса и метаболического контроля у детей с недостаточностью LCHAD или TFP [134].
При дефектах окисления длинноцепочечных жирных кислот следует ограничить потребление длинноцепочечных жиров (вычисляемое как процентная доля суммарной энергии, получаемой из жиров). При ограничении длинноцепочечных жирных кислот в рационе необходимы добавки незаменимых жирных кислот для покрытия суточной потребности в жирах (Приложение А 8). Для поддержания оптимального соотношения омега-3 и омега-6 жирных кислот предпочтительны масло грецкого ореха, соевое масло или масло ростков пшеницы [102].
3.2.7 Рекомендации по рациону питания при дефиците VLCAD
В настоящее время не существует научно-обоснованных рекомендаций по ведению пациентов с нарушениями окисления длинноцепочечных жирных кислот [62]. Гетерогенность клинических фенотипов является хорошо установленным фактом [5], что с введением в практику скрининга новорожденных также подразумевает случаи бессимптомного течения болезни [6]. Помимо этого, молекулярная гетерогенность затрудняет прогнозирование степени тяжести заболевания или исхода на момент проведения скрининга новорожденных. Выяснение вопроса о том, необходимо ли при легком фенотипе снижение в рационе доли длинноцепочечных жиров, или достаточно просто ограничиться регулярным кормлением и добавками MCT, особенно при повышенной потребности в калориях, требует более продолжительного наблюдения [66].
В зависимости от состояния ребенка энтеральное питание осуществляется через рот, а также через зонд или гастростому, расчет проводится строго индивидуально.
- Рекомендуется продолжать кормление грудным молоком или адаптированной детской смесью с добавлением специализированной низкожировой смеси с ограничением LCT из расчета 50/50 пациентам с бессимптомным вариантом VLCAD, если результаты стандартных лабораторных исследований, таких как определение активности креатинфосфокиназы в крови, активности аланинамитрансферазы в крови, аспартатаминотрансферазы в крови, глюкозы в крови находятся в пределах нормы [66].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
- Рекомендовано младенцам и более взрослым детям с бессимптомным вариантом VLCAD снижение содержания жиров в рационе до 30–40% от общей калорийности из них 10-15% энергии за счет МСТ с целью предотвращения развития метаболического криза [66].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: некоторые новорожденные с бессимптомным дефицитом VLCAD, у которых профили ацилкарнитинов полностью пришли в норму после первой недели жизни, не получали диету с низким содержанием жиров и модифицированными жирами в первые месяцы жизни, и у них до сих пор не проявились VLCAD-ассоциированные клинические симптомы. Учитывая молекулярную гетерогенность при VLCADD, очень трудно установить четкую корреляцию генотип-фенотип и спрогнозировать клиническое течение болезни. Однако мутация p.V243A, как минимум, одна копия которой обнаруживается у многих бессимптомных пациентов, выявляемых при скрининге новорожденных, как правило, ассоциирована с мягким фенотипом. У пациентов с этой мутацией может развиваться гипогликемия при тяжелых нарушениях здоровья или миопатия при сильной физической нагрузке, но возможно и бессимптомное течение.
Для пациентов, у которых развиваются только связанные с физической нагрузкой миопатические симптомы в более позднем возрасте, обогащение рациона MCT (особенно перед физической нагрузкой) может играть важную роль, а необходимость в ограничении LCT может отсутствовать.
- Рекомендуется при наличии симптомов у пациентов с дефицитом VLCAD диета с пониженным содержанием LCFA и добавление MCT жиров для предотвращения рабдомиолиза и метболической декомпенсации [61, 66].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: при наличии симптомов содержание общих жиров 25-30% от общей калорийности, из них 20-25% за счет МСТ и 5-10% за счет LCT (Приложение А15) [61].
Поскольку клинические симптомы при долгосрочном наблюдении у пациентов с дефицитом VLCAD представлены преимущественно эпизодической мышечной болью и рабдомиолизом, что указывает на недостаток мышечной энергии, рацион необходимо обогащать MCT, которые должны составлять 20% от общей калорийности (Таблица 3). Пациентам с мышечной слабостью и болью при физических нагрузках помогает повышение потребления MCT (или углеводов) непосредственно перед более интенсивной физической нагрузкой (напр., в дозировке 0,25–0,5 г MCT/кг).
3.2.8 Рекомендации по лечению пациентов с ГА2
- Рекомендуется пациентам с установленным диагнозом ГА2 соблюдение диеты с низким содержанием жиров (20-25% энергии), низким содержанием белка для снижения избыточного потребления изолейцина, лейцина, лизина, триптофана и валина [103, 111].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
- Не рекомендуется пациентам с установленным диагнозом ГА2 назначение МСТ [104].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
- Рекомендуется во время кризов пациентам с установленным диагнозом ГА2 парентеральное введение декстрозы** для купирования гипогликемии и подавления липолизиса, натрия бензоат (биологически активная добавка) для купирования гипераммониемии [111].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: детальное описание приведено в 3.1.2 Рекомендации по лечению пациентов с FAOD в период метаболической декомпенсации
3.3 Рекомендации по применению отдельных лекарственных препаратов
Кроме применения диетотерапии при нарушениях окисления жирных кислот также необходимо применение ряда лекарственных препаратов и витаминов, чтобы компенсировать дефицит незаменимых жирных кислот в организме, карнитина и жирорастворимых витаминов.
3.3.1 Рекомендации по применению отдельных лекарственных препаратов, содержащих левокарнитин
- Рекомендуется избегать назначения Левокарнитина в период криза при дефектах окисления длинноцепочечных жирных кислот, особенно в виде внутривенных инфузий для предотвращения развития рабдомиолиза, неврологических нарушений и аритмии [63, 142, 132].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: Левоакарнитин является основной терапией пациентов при первичной недостаточности карнитина. Также при низких значения карнитина в крови может использоваться при других формах FAOD с целью усиления связывания метаболитов жирных кислот и выведения их с мочой. Однако использование левокарнитина при FAOD является спорным, т.к. доказательства его эффективности при вторичном дефиците карнитина отсутствуют. При недостаточности TFP и LCHAD добавки с Левокарнитином могут индуцировать дальнейшую продукцию 3-гидроксиацилкарнитинов, способных вызывать необратимые невропатические симптомы и аритмию. Исследования на мышах с моделью дефицита VLCAD также показали, что нехватка карнитина вызывает его эндогенный биосинтез), который, однако, подавляется при экзогенном введении Левокарнитина. У многих пациентов с дефектами окисления жирных кислот, не получающих добавок Левокарнитина, концентрации свободного карнитина в плазме также варьируют от низких до нормальных, что позволяет говорить и об эндогенном биосинтезе карнитина у человека. Таким образом, добавки, содержащие Левокарнитин, должны назначаться с осторожностью, особенно, когда речь идет о недостаточности TFP и LCHAD. Существует консенсусное мнение, согласно которому следует отказаться, в частности, от внутривенного введения Левокарнитина при острых метаболических нарушениях, поскольку в таких ситуациях повышается продукция потенциально токсичных ацилкарнитинов, что приводило к внезапной смерти пациентов.
3.3.2 Рекомендации по применению незаменимых жирных кислот
Пациенты с FAOD, находящиеся на низкожировой диете подвержены высокому риску дефицита жирорастворимых витаминов. Важно избегать дефицита незаменимых жирных кислот, и большая часть потребления длинноцепочечных жиров должна поступать из масел, богатых незаменимыми жирными кислотами. Для удовлетворения потребностей в незаменимых жирных кислотах может потребоваться добавление специальных масел, таких как ореховое или льняное масло (Приложение Г 13) [105].
- Рекомендуется пациентам с недостаточностью МТР и LCHAD, назначать биологическую активную добавку в виде докозагексаеновой кислоты (DHA), играющей важную роль в формировании функций нервной и иммунной систем, а также зрительного анализатора, в дозировке 60 -65 мг/день детям с весом тела <20 кг и в дозировке 130мг в день – детям с весом тела >20 кг для поддержания функций нервной и иммунной системы [105, 137, 157].
Уровень убедительности доказательств B (уровень достоверности рекомендации – 3).
Комментарии: Линолевая кислота и α-линоленовая кислота являются предшественниками для эндогенного синтеза более длинных жирных кислот, таких как эйкозапентаеновая кислота (EPA) и докозагексаеновая кислота (DHA), которые имеют решающее значение для формирования функций нервной, иммунной систем и зрительного анализатора [105]. Обеспечить оптимальную концентрацию этих жирных кислот в плазме только с помощью применения пищевых масел сложно, поэтому пациентам требуется добавление их в чистом виде для достижения нормального уровня. Считается, что добавки DHA могут стабилизировать, и затормозить развитие пигментной дегенерации сетчатки при FAOD. Согласно проведенному исследованию [157], оптимальная диетотерапия, назначаемая на основании низких концентраций 3-гидроксиацилкарнитина и высоких концентраций DHA в плазме, позволяла сохранять функцию сетчатки и остроту зрения у детей с недостаточностью LCHAD или МТP.
- В межприступный период для пациентов с FAOD находящихся на диете с ограничением длинноцепочечных жиров рекомендовано добавлять натуральные растительные масла (кокосовое масло, масло грецкого ореха) для восполнения недостатка жирных кислот [105].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: Соотношение незаменимых жирных кислот в различных маслах приведено в Приложении А16.
- Рекомендовано назначение незаменимых жирных кислот пациентам с FAOD на фоне низкожировой диетотерапии с целью компенсации их недостаточности [11-14, 105].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: В приложении Г 8 приведены основные рекомендации по назначению незаменимых жирных кислот при диете с низким содержанием жиров.
3.3.3 Рекомендации по применению среднецепочечных триглицеридов
- Рекомендовано в качестве основных пищевых источников среднецепочечных жирных кислот использовать 50% эмульсию MCT в питании пациентов с недостаточности TFP и LCHAD, и VLCAD для предотвращения развития мышечной симптоматики [112, 149].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
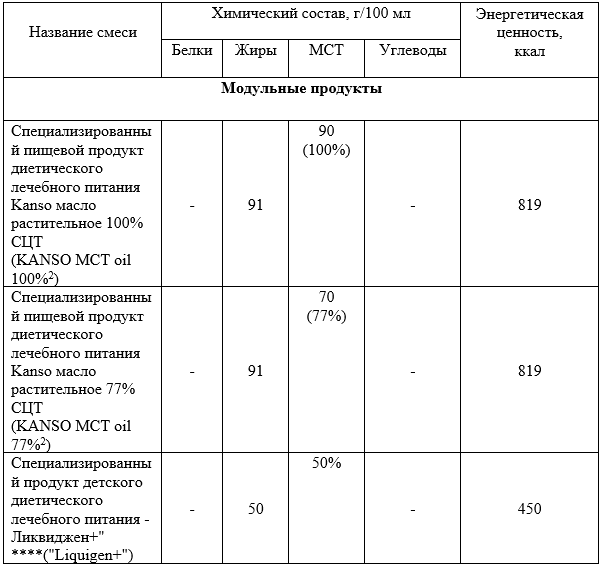
Комментарии: Содержание MCT в различных продуктах лечебного питания приведено в Приложении А17. Эпизодические мышечные симптомы, например, рабдомиолиз, патогенетически связывают с дефицитом энергии, и их можно преодолеть с помощью углеводов или MCT. В частности, по имеющимся данным, связанный с физической активностью рабдомиолиз можно предотвратить путем приема достаточного количества MCT непосредственно перед нагрузкой
- Не рекомендуется назначение пациентам с MCAD триглицеридов со средней длиной цепи (MCT) с целью предотвращения ухудшения состояния пациента. развития метаболического криза [105].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: Жирные кислоты со средней длиной цепи могут проникать в митохондрии, не будучи связанными с карнитином, минуя этап, на котором обычно регулируется окисление жирных кислот. Поэтому прием МСТ может привести к более серьезным нарушениям обмена веществ, чем при использовании длинноцепочечных жиров.
3.3.4 Рекомендации по применению витаминов
- Рекомендуется пациентам с FAOD назначение витаминов группы В и жирорастворимых витаминов А, D, Е в возрастных дозировках с целью предотвращения их дефицита [5,12, 105].Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: Витамины назначают в возрастных профилактических дозах курсами по 2-3 месяца.
- Рекомендуется всем пациентам с биохимическими нарушениями, характерными для ГА2 назначение (биологической активной добавки) рибофлавина [106, 111, 135, 136].Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: Сходный метаболический профиль может наблюдаться при дефекте транспортера рибофлавина, поэтому назначение рибофлавина (биологическая активная добавка) per os в дозе 100-200 мг/день позволяет полностью купировать клинические и биохимические симптомы болезни.
3. 4 Симптоматическое лечение
- Не рекомендуется применять препараты вальпроевой кислоты** в случае необходимости назначения противоэпилептической терапии с целью предотвращения развития печеночной недостаточности [5,12, 145].
Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
Комментарии: У пациентов с дефектами митохондриального β-окисления жирных кислот применение вальпроевой кислоты** может привести к развитию тяжелой печеночной недостаточности.
3. 5 Хирургическое лечение
При возникновении показаний для хирургических вмешательств рекомендуется коллегиальное принятие решения о тактике его проведения [5,12].